Article Text

Abstract
Objectives To evaluate in a multicentric Italian cohort of patients with psoriatic arthritis (PsA) on secukinumab followed for 24 months: (1) the long-term effectiveness and safety of secukinumab, (2) the drug retention rate and minimal disease activity (MDA), (3) differences in the outcomes according to the biological treatment line: biologic-naïve patients (group A) versus multifailure (group B) patients.
Methods Consecutive patients with PsA receiving secukinumab were evaluated prospectively. Disease characteristics, previous/ongoing treatments, comorbidities and follow-up duration were collected. Disease activity/functional/clinimetric scores and biochemical values were recorded at baseline (T0), 6(T6), 12(T12) and 24(T24) months. Effectiveness was evaluated overtime with descriptive statistics; multivariate Cox and logistic regression models were used to evaluate predictors of drug-discontinuation and MDA at T6. Infections and adverse events were recorded.
Results 608 patients (41.28% men; mean (SD) age 52.78 (11.33)) were enrolled; secukinumab was prescribed as first-line biological treatment in 227 (37.34%) patients, as second (or more)-line biological treatment in 381 (62.66%). Effectiveness of secukinumab was shown with an improvement in several outcomes, such as Ankylosing Spondylitis Disease Activity Score (T0=3.26 (0.88) vs T24=1.60 (0.69) ;p=0.02) and Disease Activity Index for Psoriatic Arthritis (T0=25.29 (11.14) vs T24=7.69 (4.51); p<0.01). At T24, group A showed lower Psoriasis Area Severity Index (p=0.04), erythrocyte sedimentation rate and C reactive protein (p=0.03 ;p=0.05) and joint count (p=0.03) compared with group B. At T24, MDA was achieved in 75.71% of group A and 70.37% of group B. Treatment was discontinued in 123 (20.23%) patients, mainly due to primary/secondary loss of effectiveness, and in 22 due to adverse events. Retention rate at T24 was 71% in the whole population, with some difference depending on secukinumab dosage (p=0.004) and gender (p=0.05).
Conclusions In a real-life clinical setting, secukimumab proved safe and effective in all PsA domains, with notable drug retention rate.
- arthritis
- psoriatic
- antirheumatic agents
- biological therapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Secukinumab (SEC) is a recent treatment for psoriatic arthritis (PsA), but real-life data are lacking.
What does this study add?
Our findings confirmed the safety and the remarkable effectiveness on all PsA domains (arthritis, enthesitis, dactylitis, spinal symptoms, psoriasis, patient-reported outcomes and inflammatory markers), over a 24-month follow-up.
The drug retention rate is considerably high, regardless of the biological treatment line.
Male gender seems to favour minimal disease activity achievement and drug persistence, while higher body mass index is associated with drug discontinuation.
How might this impact on clinical practice?
This study support the effectiveness of SEC, which seems also a valid option for monotherapy and in multifailure patients; the safety of SEC allows its use in patients with comorbidities, in particular cardiovascular conditions and metabolic syndrome.
Introduction
Psoriatic arthritis (PsA) is a chronic, inflammatory disease characterised by peripheral arthritis, axial disease, dactylitis, enthesitis and skin and nail psoriasis.1 2 PsA can negatively affect patients’ daily functioning and quality of life, due to permanent joint damage and disability.3 The reported prevalence of PsA in the general population is up to 1%, and the disease affects around 30%–40% (range 6%–42%) of patients with psoriasis.4–6
Biologic therapies, such as antitumour necrosis factor (TNF), anti-interleukin (IL)-12/23 and anti-IL-17A antibodies, are recommended for the treatment of PsA in patients who respond inadequately to first-line treatment with non-steroidal anti-inflammatory drugs (NSAIDs) and/or conventional synthetic disease-modifying antirheumatic drugs (csDMARDs).7–10 Proinflammatory cytokine IL-17A, the main effector cytokine of Th17 cells, mediates multiple biological functions in PsA, resulting in joint and entheseal inflammation and structural damage.11
IL-17A stimulates keratinocytes to release inflammatory molecules that mediate skin damage in cutaneous psoriatic lesions and recruit neutrophils, monocytes, Th17 cells and other cell types on site.12 IL-17 is also produced in arthritic joints13 14 and in entheseal structures15 through local stimulation of IL-22 and IL-23.
Recommendations from the EULAR16 and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA)17 or the Italian Society of Rheumatology (SIR) guidelines18 recognise targeting IL-17A as a therapeutic strategy to manage all the main clinical manifestations of PsA. Secukinumab (SEC) is a fully human monoclonal antibody that selectively binds to and neutralises IL-17A. It has been approved for use as a subcutaneous injection for the treatment of moderate-to-severe psoriasis, PsA and ankylosing spondylitis (AS).19 20 The long-term extension of pivotal trials showed the sustainable effectiveness and safety for SEC up to 5 years in PsA; some dedicated randomised controlled trials (RCTs), in fact, demonstrated the effectiveness of this drug in reducing some disease manifestations, such as skin lesions, arthritis, enthesitis and dactylitis, thus leading to significant improvement in mobility, radiographic progression, and quality-of-life parameters.21–25
Despite the presence of a robust positive evidence from the RCTs on the use of SEC, it is important to demonstrate the effectiveness in real life. While RCTs assess the drug effectiveness and safety, daily practice may often be different, since some patients may be excluded or under-represented in clinical trials because of significant comorbidities or previous multiple therapy failures.21–26 Thus, real-life data on the wide spectrum of patients affected by the disease are needed, in order to evaluate the effectiveness and safety of this biologic drug. Until now, few prospective studies presented real-life data on the treatment of PsA with SEC.27–31 In fact, the evidence of the effectiveness and safety of SEC in the real-world setting has been mainly generated from independent and company-sponsored registries and postmarketing phase IV studies.32–34
The current prospective observational study aimed to: (i) evaluate the long-term effectiveness of SEC in the management of PsA manifestations, including joint and skin symptoms, in a real-life clinical setting for a 24-month follow-up period; (ii) identify the differences in the outcome measures of effectiveness according to the biological treatment line; (iii) analyse MDA according to dosage administered (150 or 300 mg/injection), biological treatment line, gender, body mass index (BMI); (iv) identify predictors of MDA and treatment discontinuation at 6 months; (v) describe any comorbidities; (vi) report any adverse events or infections and (vii) evaluate drug survival at 24 months.
Materials and methods
Patients
The present longitudinal study included consecutive patients with PsA who underwent the treatment with SEC from September 2016 to March 2020 in 12 Italian Rheumatology centres. The study was supported by the Italian SIR ‘Spondyloartritis and Psoriatic Arthritis study group—A. Spadaro’.
Patients started treatment with SEC according to EULAR and/or GRAPPA, and/or the Italian SIR guidelines.16–18 Inclusion criteria were: age ≥18 years and a diagnosis of PsA for >6 months, in accordance with the Classification for Psoriatic Arthritis criteria,35 and the indication to begin SEC treatment for a moderate or severe disease. The concomitant use of csDMARDs was allowed, provided the dosage had been stable over the previous 3-month period. Although the discontinuation or reduction in csDMARD dosage was permitted during the study period, if the patient’s clinical situation was favourable, increasing the dose was not allowed. NSAIDs and low-dose corticosteroids (daily dose of 10 mg or less of prednisone or equivalent) were permitted. Intra-articular corticosteroid injections were not allowed.
We excluded patients with PsA who showed any of the following: a history of malignancy (during the last 5 years) and/or systemic infections, active infectious disorders (including active tuberculosis), congestive heart failure and demyelinating diseases.
Screening and evaluation
Patients underwent a series of screening tests before enrolment and starting treatment, in compliance with the European guidelines.16–18 Screening included chest X-ray, laboratory tests (screening for HIV and hepatitis B and C viruses), the Mantoux TB skin test or Quantiferon TB Gold test and a pregnancy test for all women of childbearing age. Patients presenting latent tuberculosis underwent a complete 6-month prophylaxis with isoniazid (300 mg/day). Patients were evaluated at baseline (T0) and after 6 (T6), 12 (T12) and 24 (T24) months of SEC treatment. Relevant anamnestic, clinical, biochemical and metrological data were collected. These included the biological treatment line, which was considered as a dichotomous variable (SEC as a first-line biological treatment/as a second-line or more biological treatment), and arthritis subtype, which was considered a categorical variable (polyarticular-monoarticular/oligoarticular-axial-only involvement of distal interphalangeal joints (DIP)-mutilating arthritis). Clinical variables—such as psoriasis, dactylitis, enthesitis and smoking status—were considered as dichotomous (yes/no) on patient enrolment. For the analyses, patients with PsA were subdivided in two groups: (a) naïve to any biologic drug (group A) and patients who had failed TNF-inhibitors (TNFi) or anti-IL12/23 agents (group B). The presence of comorbidities and concomitant therapies were investigated (yes/no) during a face-to-face interview at one of the scheduled assessment visits and by reviewing the patients’ medical records. Information on previous therapies with biological DMARDs (bDMARDs) (and in particular the line/s of treatment used) and concomitant csDMARDs (methotrexate, leflunomide, sulfasalazine, hydroxychloroquine and ciclosporin) or corticosteroid therapy or NSAIDs were recorded at baseline and throughout the study.
Effectiveness measures and outcomes
Relevant patient-reported outcomes (PROs),36 such as the Visual Analogue Scale of pain (VAS-pain) and global health (VAS-gh), Health Assessment Questionnaires modified for spondyloarthritis (HAQ-S), Bath Ankylosing Spondylitis Functional Index (BASFI) and Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) were collected. The clinical evaluation, made by an experienced rheumatologist and an experienced dermatologist (the same assessor at each time point), included the Psoriasis Area Severity Index (PASI), the assessment of the presence of psoriatic onychopathy and dactylitis (yes/no), joint tenderness (in 68 joints) and swollen joint count (in 66 joints) as well as the Disease Activity in Psoriatic Arthritis (DAPSA) score and the Ankylosing Spondylitis disease activity score (ASDAS).36 Enthesitis was assessed using the Leeds Enthesitis Index (LEI), and dactylitis was expressed as the number of digits involved. The evaluation of the fulfilment of minimal disease activity (MDA) was carried out in the study population in accordance with Coates’ composite measure: those who fulfilled five out of the seven criteria were considered to have experienced MDA.37 MDA was also calculated after the subdivision of all population in two groups, according to the lines of DMARDs treatment (bio-naïve vs non-bio-naïve patients), gender, BMI (overweight vs normal weight) and dosage of SEC (150 vs 300 mg/injection).
Biochemical acute phase reactants (erythrocyte sedimentation rate (ESR) and the C reactive protein (CRP)) values were determined and analysed. Our normal lab ranges were as follows: ESR 0–28 mm/hour female, 2–38 mm/hour male; CRP 0–6 mg/L. Metrological indexes such as height, weight and BMI were evaluated.
Administration of SEC
All patients were treated with SEC, administered subcutaneously at a dosage of 150 or 300 mg as needed—according to the decision of the treating rheumatologist and the national registration indications of the drug—for psoriasis or multidrug failure at weeks 0, 1, 2, 3, 4 and every 4 weeks thereafter in accordance with the manufacturer’s instructions.38 Patients were taught to perform self-injections. The drug’s safety was evaluated by assessing adverse events and through standard laboratory testing, and the patients’ vital signs were evaluated and recorded throughout the study, and for a 60-day period following the last treatment. Drug survival was calculated as the number of days in which the patient remained on therapy. The start date was the day the first dose was taken and the stop date was the day the treatment was interrupted. Temporary interruptions (eg, due to infections or surgery ≤3 months before) were acceptable according to our study protocol. Primary inefficacy was considered as ‘no satisfactory improvement of clinical manifestations during the first 6 months of SEC treatment according to physician’s judgement’. A secondary inefficacy was defined as ‘reappearance of PsA manifestations for at least 4 weeks leading to SEC withdrawal despite a previous global response lasting at least 6 months’.
Statistical analysis
Data were expressed as frequencies and percentages for categorical variables, and as mean±SD for continuous variables. Patients’ characteristics were compared between group A and group B using the χ2 test or the Fisher’s exact test for categorical variables and the t-test or the Wilcoxon rank test for continuous variables, based on data distribution. Effectiveness data on arthritis, psoriasis, enthesitis and dactylitis were compared between T0 and T24 with the χ2 test or the Wilcoxon rank test, as appropriate.
The time-to-event analysis was performed according to the Kaplan-Meier method. Survival curves were compared—by log-rank test—between: (a) lines of bDMARDs treatment (bio-naïve vs non-bio-naïve patients), (b) gender, (c) BMI (overweight vs normal weight) and (d) dosage of SEC (150 vs 300 mg/injection).
A logistic regression analysis was carried out in order to identify predictors of MDA at T6, and a Cox regression model was built to identify predictors of time-to-SEC discontinuation. In both cases, baseline independent factors which, based on literature data,31 39 could be expected to have an effect on short-term MDA achievement or on drug discontinuation, were considered. Results were expressed as OR for logistic regression and HR for Cox regression, together with their 95% CIs. All statistical analyses were carried out with the SPSS V.13.0 software (SSPS, Illinois, USA). Two-tailed p values ≤0.05 were considered statistically significant.
Results
Patients features
Six hundred and eight patients with PsA (41.28% men; mean age 52.78 (11.33)) were enrolled; mean disease duration was 9.49 (6.85) years, and mean treatment duration was 18.77 (11.17) months. SEC was prescribed as first-line biological treatment in 227 (37.34%) patients (group A) and as second (or more)-line biological treatment in 381 (62.67%) patients (group B); 352 patients (57.89%) were in monotherapy. At baseline, 274 (45.97%) and 334 (54.93%) patients were receiving SEC 150 mg/injection and SEC 300 mg/injection, respectively. The patients’ clinical and laboratory baseline (T0) characteristics, such as concomitant treatments, are summarised in table 1. Polyarthritis was present as a prominent manifestation in 59.70% of the cases; asymmetric oligoarthritis or monoarthritis in 26.81%; axial involvement with sacroiliitis and/or spondylitis was present in 24.01% and enthesitis in 43.42% of the patients. Erosive disease was present at baseline in 28.62% of the patients, and three cases of arthritis mutilans and nine cases of prominent DIP involvement were recorded. The following extra-articular manifestations were recorded: inflammatory bowel disease (IBD) (1.81%, n=11) and uveitis (4.11%, n=25) in remission.
Baseline characteristics of 608 patients with PsA treated with SEC in the period September 2016–March 2020
At T0, 256 (42.11%) patients were receiving concomitant csDMARDs at a stable dosage for at least 3 months, 162 (26.64%) were taking glucocorticoids and 314 (51.64%) were on NSAIDs.
At T0, group B (compared with group A) had a more erosive and polyarticular pattern, a more frequent axial involvement, a longer disease and psoriasis duration, a greater prevalence of dactylitis, a higher glucocorticoids intake, a higher prevalence of extra-articular features, such as IBD and uveitis. No significant difference was observed for enthesitis and the severity of psoriasis and the clinical and functional parameters (table 1). Few clinical and anthropometric features varied between males and females: at baseline males presented a higher prevalence of dactylitis and psoriasis, a higher PASI score, while females presented a higher ESR value and tender joint count. No significant differences were found about disease activity indexes values (ie, DAPSA) and type and prevalence of concomitant cDMARDs (online supplemental table 1).
Supplemental material
Therapy effectiveness
Of all 608 patients with PsA, 502 (82.57%) were evaluated at T6, 406 (66.78%) at T12 and 205 (33.72%) at T24.
The whole population achieved a significant decrease in tender/swollen joints (TJ/SJ), VAS-pain, VAS-gh, PASI, LEI, number of dactylitis, HAQ-S, BASDAI, BASFI, CRP (table 2). An improvement in ASDAS (T0=3.26 (0.88) vs T24=1.60 (0.69); p=0.02) and in DAPSA (T0=25.29 (11.14) vs T24=7.69 (4.51); p<0.01) was also noted.
Clinical, functional, disease activity and serological parameters of all (n=608) patients with PsA during 24 months-follow up
During the 24 months follow-up, a significant reduction was noted in the number of patients with active joint count (TJ at T0=94.57% (n=575) vs TJ at T24=43.92% (n=90); SJ at T0=66.12% (n=402) vs TJ at T24=16.10% (n=33); p<0.01); enthesitis (at T0=43.42% (n=264) vs at T24=17.56% (n=36); p<0.01); dactylitis (at T0=15.95% (n=97) vs at T24=9.75% (n=20); p<0.01) and psoriasis (at T0=68.75% (n=418) vs at T24=15.12% (n=31); p<0.01).
At T24, group A showed better physical functioning and lower inflammatory activity compared with group B (VAS-pain A vs B=1.62 (1.72) vs 3.00 (2.45) (p=0.04); VAS-gh A vs B=1.34 (1.66) vs 3.01 (2.40) (p=0.03); ESR A vs B=11.98 (10.44) vs 16.10 (13.02) (p=0.03); CRP A vs B=2.12 (3.06) vs 4.04 (6.03) mg/L (p=0.05)), while group B maintained a higher TJ count and PASI score than group A (TJ A vs B=0.54 (1.02) vs 2.11 (3.57) (p=0.03); PASI A vs B=0.23 (0.55) vs 1.14 (3.76) (p=0.04)) (table 3).
Clinical, functional, disease activity and serological parameters of naïve (n=227) and biological agents failure (n=381) in patients with PsA during 24 months follow up
After T24 of treatment, 75.71% of group A and 70.37% of group B reached MDA, respectively (p=0.04) (figure 1A). We also evaluated the proportion of achievements of MDA according to gender, BMI and SEC dosage. As shown in figure 1B, patients receiving SEC 150 mg/injection achieved MDA in a higher proportion compared with those who received SEC 300 mg/injection (MDA SEC 150 vs SEC 300=81.11% vs 70.37% (p=0.02)). No differences were observed in the proportion of MDA achievement in relation to gender (figure 1C) or BMI (figure 1D).

Minimal disease activity (MDA) (percentage, %) of overall population and after their subdivision in two groups, according to the lines of the disease-modifying antirheumatic drugs treatment (group A, or bio-naïve vs group B, or non-bio-naïve patients) (A), dosage of secukinumab (150 vs 300 mg/injection) (B), gender (C) and body mass index (BMI) (overweight vs normal weight) (D). ns, not statistically significant; PsA, psoriatic arthritis.
The proportion of patients on csDMARDs was higher at T0 (42.11%, n=256) than at T6 (38.84%, n=195), at T12 (36.20%, n=147) and at T24 (27.32%, n=56), as was the proportion of patients treated with glucocorticoids, which were 26.64% (n=162) at T0, 12.55% (n=63) at T6, 10.34% (n=42) at T12% and 6.34% (n=13) at T24. A high reduction in NSAIDs intake was observed from T0 (51.64%, n=314) to T6 (31.08%, n=156), T12 (24.14%, n=98) and T24 (20.9%, n=43). During the follow-up of 24 months, it was only found a more less reduction of ESR value in females, while all other clinical/functional/disease activity indexes improved similarly between males and females (online supplemental table 2).
Supplemental material
Drug survival
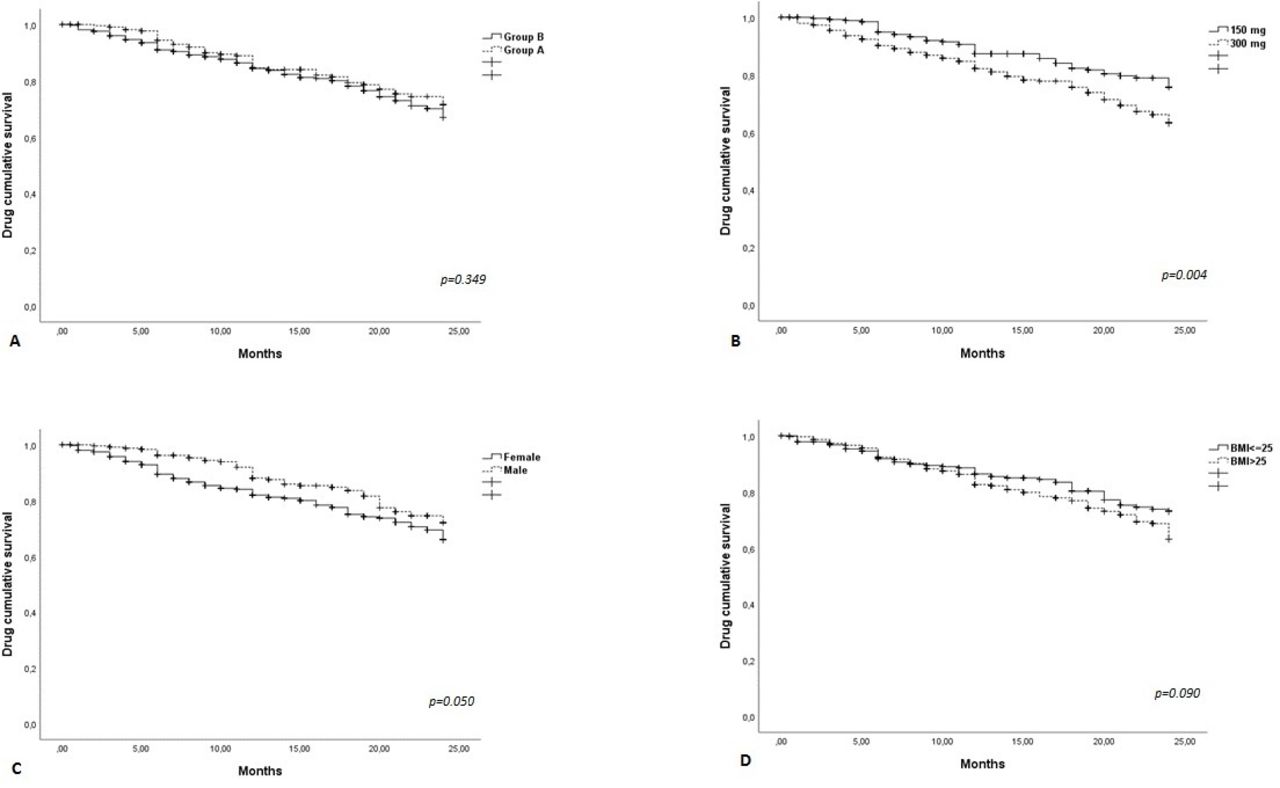
The retention rate at T24 was good (71%) in the whole population (figure 2A–D). The Kaplan-Meier curves did not display any differences between group A and group B (log-rank 0.88; p=0.349) and between subjects with BMI ≤25 and those with BMI >25 (log-rank 2.873; p=0.090), while a significant difference was found between patients receiving the 150 mg/injection dosage and those receiving the 300 mg/injection dosage (log-rank 8.14; p=0.004)—with patient in the 150 mg group having a higher survival—and between the male and female populations (log-rank 3.75; p=0.050).

{kind=link}
{kind=link}
Drug survival in the overall population and after their subdivision in two groups, according to the lines of the disease-modifying antirheumatic drugs treatment (group A, or bio-naïve vs group B, or non-bio-naïve patients) (A), dosage of secukinumab (150 vs 300 mg/injection) (B), gender (C) and body mass index (BMI) (overweight vs normal weight) (D).
Predictors of effectiveness
A multivariable regression analysis was performed to identify any baseline predictors of achievement of MDA at T6. As shown in table 4, male gender was associated with a higher chance of MDA achievement at T6, while a higher baseline DAPSA and the use of csDMARDs were negatively associated with the outcome. Age, active psoriasis, BMI, a 300 mg/injection dosage and the line of biological treatment were not independently associated with MDA at T6.
Multivariable logistic regression model with MDA at T6 as outcome and Cox regression model with time-to-SEC withdrawal as outcome
A Cox regression analysis was performed to identify predictors of time-to-SEC discontinuation. The variables that were independently associated with a higher chance of SEC discontinuation were: female gender, 300 mg/injection dosage and higher BMI (table 4).
Comorbidities
The most frequently observed comorbidities were: metabolic syndrome (MetS) (12.34%, n=75), hypertension (39.14%, n=238), ischaemic heart disease (7.57%, n=46), dyslipidaemia (26.64%, n=162), hyperuricaemia (16.78%, n=102), type II diabetes (9.70%, n=59), gastritis, gastric ulcer or dyspeptic disorders (12.50%, n=76), liver disease (eg, steatosis) 11.35%, n=69), pneumopathies (6.74%, n=41), thyroid disorder (12.82%, n=78), osteoporosis (8.39%, n=51), kidney failure (2.14%, n=13), depression (11.84%, n=72), fibromyalgia (16.78%, n=102), neurological disorders (such as neuropathy) (5.92%, n=36), positive Mantoux RB skin test or Quantiferon TB Gold test (6.58%, n=40) without active tuberculous disease, previous hepatitis B (8,22%, n=50), previous hepatitis C (2.30%, n=14), previous eradicated cancer (5.10%, n=31). The frequency of these comorbidities were described in both group A and group B (table 5). A higher prevalence of cardiovascular pathologies, diabetes, hepatic steatosis and MetS in the group B was found.
Comorbidities of naïve (n=227) and biological agents failure (n=381) in patients with PsA
Safety and discontinuation
Reasons of discontinuation
SEC was found to be safe and well tolerated.
One hundred twenty-three patients (20.23%) stopped the treatment during the follow-up, mainly because of primary and secondary loss of effectiveness (46 and 77, respectively). Fifteen patients dropped out from the observational study. Only 22 patients suspended SEC because of adverse events (6 for reactions at the injection site, 1 for leukopenia-neutropenia, 1 for hypertransaminasaemia, 2 for dyspnoea, 1 for multiple sclerosis, 6 for severe recurrent infections, 5 due to the onset of new cancer: 1 prostate cancer; 1 acute myeloid leukaemia; 1 breast cancer, 1 renal cancer, 1 pulmonary adenocarcinoma). Liver and renal function was monitored in all patients during the study period, and only two of the patients presented abnormal values.
Infections
A low number of episodes of mild infections (72) occurred during the study period (27 respiratory tract infections; 10 oral or vaginal candidiasis; 3 herpetic labial infections; 3 herpes zoster; 7 gastroenteritis or diverticulitis; 22 urinary tract infections); all resolved following oral antimicrobial treatment, without hospitalisation or drug discontinuation. Six patients presented severe infections, which caused them to suspend the SEC treatment (two bronchopneumonia and one erysipelas with sepsis from St. Aureus with hospitalisation and three recurrent candidiasis). No difference between dosage/gender/BMI/group A versus group B in terms of safety (data not shown) was observed.
Discussion
In this prospective multicentric study in 608 patients with moderate-to-severe PsA, SEC—a monoclonal antibody that selectively binds to and neutralises IL-17A—was effective (for both musculoskeletal symptoms and cutaneous psoriasis), safe and well tolerated. In addition, over three-quarters of patients were able to achieve MDA at 6 months, and the majority of them remained on treatment after 24 months.
At T6, T12 and T24, SEC reduced all the clinical and inflammatory indexes analysed, in addition to PROs. We also observed a relevant improvement in disease activity parameters, with a significant reduction of the DAPSA, BASDAI and ASDAS scores. These findings well demonstrate the effectiveness of SEC in PsA, in accordance with the RCTs. In particular, in the FUTURE 1 and 2 studies, which assessed the 5-year effectiveness and safety of SEC, about 66% of patients achieved ACR20 (lower percentages were reached for ACR50 and ACR70) and the clinical response was sustained throughout 5 years.20–22 25
The effectiveness of SEC in enthesitis was known from the pooled data of the FUTURE 2 and 3 studies, which showed the resolution of enthesitis (as assessed by LEI) in over half of the patients at week 16, and in the vast majority of them at week 104.11 40 41 In our population, 43.42% of patients with PsA showed entheseal involvement. Mean LEI significantly decreased throughout the study period, and a higher—although non-significant—response at T24 was observed in group A compared with group B (table 3). This result confirmed that SEC was effective in reducing the frequency of enthesitis24 25 31 and could be indicated in patients with PsA with prevalent entheseal involvement. Moreover, in FUTURE 2, almost half of patients achieved the resolution of dactylitis.42 Similarly, in our study a significant reduction in the prevalence of dactylitis throughout the 2-year follow-up was observed (15.95% at T0 and 9.75% at T24). Therefore, SEC showed effectiveness in both enthesitis and dactylitis.
The optimal response to SEC in psoriasis is widely reported in the RTCs. Phase III RCTs show that in patients with moderate-to-severe psoriasis SEC provides significant rates of skin clearance, which is sustained up to 5 years,43–46 especially in bio-naïve patients,47 48 as well as a favourable safety profile. Of the entire population of 608 patients, 68.75% presented active psoriasis at baseline, while at T24 only 15.12% of patients showed signs of active psoriasis, with a significantly reduction in PASI (p<0.01), which was more marked in group A than in group B (0.23 vs 1.14, respectively). Meaningful effectiveness was also observed in challenging-to-treat variants of psoriasis, including nail, scalp and palmoplantar pustolosis.19 Danish registry DERMBIO reported a rate of persistence on adherence to the treatment with SEC at 3 years of around 80% among patients naïve to previous biologics, and between 30% and 50% among non-responders to previous anti-TNF agents.49 Therefore, SEC appears to be more effective than anti-TNF-α in severe and/or extensive psoriasis. We also observed a remarkable effectiveness in 24% patients with axial involvement, measured by the significant reduction of ASDAS and BASDAI scores. These findings were in line with Managing AXIal Manifestations in psorIatic arthritis with SEcukinumab, the first RCT who evaluated the efficacy of SEC specifically in the management of the axial manifestations of PsA: SEC 300 mg and 150 mg demonstrated significant improvements across the clinical (ASDAS, ASAS20/40 and BASDAI50) and imaging (Berlin MRI Score) end points at week 12, which were sustained through week 52.50
Our results confirmed the effectiveness of SEC in multiple aspects of PsA, in accordance with the need to treat the heterogeneous clinical phenotypes. The updated PsA core domains, defined by the GRAPPA/OMERACT, underline the need to obtain a good treatment response in arthritis, enthesitis, dactylitis and spine involvement, skin disease activity (psoriasis and nail involvement), pain, patient global assessment, physical function, fatigue, health-related quality of life and systemic inflammation.51 The findings of our study highlighted that SEC was effective across all GRAPPA-OMERACT PsA core domains. These results are similar to those reported in the pooled analysis on 2049 patients in the FUTURE 2–5 trials, which suggested that PsA can benefit from SEC across the whole clinical phenotype spectrum commonly encountered in this disease.52 Similarly, at EULAR 2019 EuroSpA—a research collaboration network among 16 registries conducted in 16 countries— presented data from >1500 patients with PsA treated with SEC, of whom 80% had biological drug experience, with a mean disease duration of 10 years. The EuroSpA research showed that the Disease Activity Score 28 CRP (DAS28CRP), Simplified Disease Activity Index (SDAI) and Disease Activity in PsA 28 (DAPSA28) remission at 6 months were achieved by 35%, 12% and 12% of patients, respectively.33 The overall retention rates for bio-naïve and non-naïve patients was 86%, with significant differences across the registries.34 53
In our study, group B (381 patients) had been previously treated with biological agents and 227 were bio-naïve (group A). An improvement in all clinical, serological and disease activity indexes and PROs were observed in both groups, with a higher response in joint count, inflammatory markers and VAS-pain in group A. Therefore, SEC appears to be effective also in non-responder patients with PsA, and can be considered as a drug of choice in patients with one or more previous bDMARDs failure.54 Moreover, a large body of evidence from phase III trials FUTURE 1 and 2 showed that SEC is a very effective treatment in patients with PsA, both naïve to previous line treatment and non-responders.11 25 In our study, the retention rate was found to be similar in both groups, A and B, regardless of the treatment line. In this context, the efficacy of SEC was independent of the line of treatment used and previous use of bDMARDs and can therefore be considered effective as first-line therapy and in multifailure patients. Data from national registries, such as the ATTRA registry, have also compared the characteristics of patients starting treatment with SEC or TNF inhibitors and the effectiveness of SEC was similar in both naïve and non-naïve patients with PsA.55
The use of combination therapy with corticosteroids or csDMARDs is still controversial in the management of PsA, and its rationale is based on evidence or expert opinion.16–18 The combined therapy may be adopted in case of inadequate control of peripheral joint manifestations. In this study, about 40% of patients were treated with csDMARDs at baseline. However, a reduction in the concomitant use of csDMARDs and a tapering of low-dosage glucocorticoids were noted during the 24-month follow-up, thus demonstrating the effectiveness of SEC also as monotherapy. This interesting observation could allow clinicians to consider SEC in patients unsuitable for the treatment with csDMARDs or glucocorticoids.
A good retention rate (71%) was observed in all our study population. Discontinuation of treatment (20.23%) was mainly due to primary and secondary effectiveness loss (46 and 77 cases, respectively).
In our patient, MDA was reached in 47.65% of cases within the first 6 months of treatment, and during the 24-month follow-up period this proportion increased up to 72.2% at T24. MDA was achieved in a high proportion of patients regardless of the treatment line, although with a slight difference between groups A and B (75.71% vs 70.37%, respectively). An important observation is that about gender influence on the achievement and maintenance of MDA. While comparable percentages of males and females (74.03% vs 70.41%) reached MDA at T24, male sex was a predictor of MDA achievement at T6. Interestingly, in literature female gender was found to be associated with poorer rates of response to bDMARDs (ie, TNFi) and with a lower probability of achieving remission, compared with males.56 In addition, a higher baseline DAPSA and concomitant csDMARDs intake were negative predictors of MDA achievement at T6. Indeed, these variables were also recognised as characteristics of severity and of an active stage of the disease, so our findings are likely explained by the fact that more severe patients might require more time to reach MDA.39 57
As far as time-to-SEC discontinuation was concerned, male sex appeared to be protective against drug discontinuation. This finding is in contrast with some studies in psoriasis, which failed to demonstrate an influence of sex on drug survival,58 59 while it is in accordance with other studies which confirmed a role of male sex in favouring biologic treatment survival.60 Moreover, also the dosage of SEC appeared to affect drug survival, which was longer with 150 mg. This can be explained by the fact that the 300 mg dose was mainly used in non-naïve patients or in severe PsA/psoriasis, where a lower response is indeed expected. Likewise, in this study overweight patients or patients with obesity having PsA showed a higher risk of SEC discontinuation: for each 1-point increase in BMI, the hazard rate of discontinuing SEC increased by 4%. This is congruent with numerous studies showing that obesity can influence the clinical response to systemic treatment, especially to targeted immunomodulators such as anti-TNF-α drugs.61–63
In our study, SEC demonstrated a good safety profile (only 22 cases leading to drug withdrawal for adverse events). A pooled safety analysis from phase III RCT also supports the favourable long-term safety of SEC in patients with psoriasis and PsA.32 The exposure-adjusted incidence rates (EAIR) for infections at 1 year were comparable across the three drug groups (secukinumab, etanercept and ustekinumab). One of the most commonly reported infections—which however does not usually lead to the interruption of IL-17 inhibitors—was mucocutaneous candidiasis.11 32 In this study, mucocutaneous candidiasis was also observed in 10 patients. Other infections were of mild-to-moderate intensity and did not lead to treatment discontinuation. The onset of IBD was not identified for up to 2 years of treatment. The real-life safety profile of SEC was therefore similar to the results of phase II/III studies.64 Forty (6.58%) patients presented positive Mantoux RB skin test or Quantiferon TB Gold test when they started treatment with SEC. None of these patients developed an active tuberculous disease during the course of treatment. No difference between dosage/gender/BMI/group A versus group B in terms of safety was observed. Even in group B which had a higher prevalence of cardiovascular diseases, diabetes and MetS (probably due to a longer history of PsA disease and side effects derived from multiple drug treatment lines, in particular steroids), few were the patients who had stopped SEC for treatment-related adverse events or infections.
A limitation of our study was the absence of imaging follow-up (X-rays, ultrasound or MRI), although imaging was often performed to confirm the diagnosis at baseline. In addition, axial PsA is distinct from axSpA and hence we did not apply stringent radiographic or MRI criteria such as those commonly used in the latter form. Furthermore, the lack of axial PsA-specific outcome measures brought on the challenge of choosing the appropriate outcome measures. It is well-recognised that there is an unmet need for axial PsA-specific outcome measures as ASAS and BASDAI, although working well in AS trials are not specific for axial inflammation in PsA. On the other hand, its strong points are the prospective study design and the large multicentre study population.
Conclusions
In a real-life clinical setting, SEC was safe and effective in PsA, as shown by a significant decrease in DAPSA and ASDAS over a 24-month follow-up. Our findings confirm its remarkable effectiveness on all PsA domains (arthritis, enthesitis, dactylitis, spine symptoms such as those on psoriasis and PROs and inflammatory markers), regardless of the biologic treatment line. SEC also seems a valid option for monotherapy, since a reduction in the concomitant use of csDMARDs and glucocorticoids was observed during follow-up. Therefore, clinicians might consider the use of SEC both in first and subsequent treatment lines, as well as in patients who are unsuitable for csDMARDs or glucocorticoids. As suggested by the significant drug retention rate, SEC was able to maintain its effectiveness over a considerable long period of treatment. Male gender seems to favour MDA achievement and drug persistence, while higher BMI is associated with drug discontinuation. The safety of SEC allows its use in patients with comorbidities, in particular cardiovascular conditions and MetS.
Acknowledgments
The authors would like to thank Italian Society of Rheumatology (SIR) “Spondyloartritis and Psoriatic Arthritis Study Group—A. Spadaro”. The authors would also like to thank the authors of ref. 31 and 39 for granting permission for their articles' reprint.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AC and RF are joint senior authors.
RR and ML contributed equally.
Collaborators Coordinators: Raffaele Scarpa, MD, PhD, Rheumatology Research Unit, Department of Clinical Medicine and Surgery, School of Medicine and Surgery, University of Naples FEDERICO II, Naples, Italy; Antonio Marchesoni, MD, PhD, Department of Rheumatology, ASST Gaetano Pini-CTO, Milan, Italy; Carlo Salvarani, MD, PhD, Rheumatology Unit, Department of Internal Medicine, Azienda USL-IRCCS, Istituto di Ricovero e Cura a Carattere Scientifico, Reggio Emilia, Italy; University of Modena and Reggio Emilia, Modena, Italy; Salvatore d’Angelo, MD, PhD, Rheumatology Institute of Lucania (IReL)—Rheumatology Department of Lucania, San Carlo Hospital of Potenza and Madonna delle Grazie Hospital of Matera, Potenza, Italy; Alberto Cauli, MD, PhD, Rheumatology Unit, Department of Medical Sciences, AOU and University of Cagliari, Cagliari, Italy; Ennio Lubrano, MD, PhD, Academic Rheumatology Unit, Dipartimento di Medicina e Scienze per la Salute "Vincenzo Tiberio", University of Molise, Campobasso, Italy; Roberta Ramonda, MD, PhD, Rheumatology Unit, Department of Medicine, University of Padova, Padova, Italy. The members of the “Spondyloartritis and Psoriatic Arthritis Study Group—A. Spadaro” who contributed to this study are as follows: Antonio Carriero, MD, PhD, Rheumatology Institute of Lucania (IReL)—Rheumatology Department of Lucania, San Carlo Hospital of Potenza and Madonna delle Grazie Hospital of Matera, Potenza, Italy; PhD Scholarship in Translational and Clinical Medicine, Department of Medicine and Health Sciences, University of Molise, Campobasso, Italy; Maria Sole Chimenti, MD, PhD, Rheumatology, Allergology and Clinical Immunology, Department of “Medicina dei Sistemi”, University of Rome “Tor Vergata”, Rome, Italy; Angelo Semeraro, MD, Rheumatology Unit, Martina Franca-ASL Taranto, Italy; Leonardo Santo, MD, Rheumatology Unit, Azienda Sanitaria Locale Barletta Andria Trani—DSS4 Barletta, Italy, Barletta-Andria-Trani, Italy; Mariagrazia Lorenzin, MD, PhD, Rheumatology Unit, Department of Medicine, University of Padova, Padova, Italy; Augusta Ortolan, MD, Rheumatology Unit, Department of Medicine, University of Padova, Padova, Italy; Andrea Doria MD, PhD, Rheumatology Unit, Department of Medicine, University of Padova, Padova, Italy; Elena Fracassi, MD, Rheumatology Unit, Department of Medicine, AOUI University of Verona, Verona, Italy; Giulia Virelli, MD, Rheumatology Unit, Department of Medicine, AOUI University of Verona, Verona, Italy; Marco Masia, MD, Rheumatology Unit, Department of Medicine, AOUI University of Verona, Verona, Italy; Rosalinda Fanizzi, MD, Rheumatology Unit, Martina Franca-ASL Taranto, Italy; Elisa Visalli, MD, Rheumatology Unit, A.O.U. Policlinico S. Marco, Catania, Italy; Giorgio Amato, MD, Rheumatology Unit, A.O.U. Policlinico S. Marco, Catania, Italy; Antonio Carletto, MD, Rheumatology Unit, Department of Medicine, AOUI University of Verona, Verona, Italy; Rosario Foti, MD, Rheumatology Unit, A.O.U. Policlinico S. Marco, Catania, Italy; Luisa Costa, MD, PhD, Rheumatology Research Unit, Department of Clinical Medicine and Surgery, School of Medicine and Surgery, University of Naples FEDERICO II, Naples, Italy; Desiati Francesca, MD, Department of Rheumatology, ASST Gaetano Pini-CTO, Milan, Italy; Giulia Lavinia Fonti, MD, Rheumatology, Allergology and Clinical Immunology, Department of “Medicina dei Sistemi”, University of Rome “Tor Vergata”, Rome, Italy; Nicolò Girolimetto, MD, Rheumatology Unit, Department of Internal Medicine, Azienda USL-IRCCS, Istituto di Ricovero e Cura a Carattere Scientifico, Reggio Emilia, Italy; Fabio Massimo Perrotta, MD, Academic Rheumatology Unit, Dipartimento di Medicina e Scienze per la Salute "Vincenzo Tiberio", University of Molise, Campobasso, Italy; Emanuela Praino, MD, Rheumatology Unit, Azienda Sanitaria Locale Barletta Andria Trani—DSS4 Barletta, Italy.
Contributors RR and AC were responsible for the study conceptualisation and design. ML and AO performed the statistical analysis. AC, MSC, RS, AM, EL, CS, AC, AS, LS, AD, EF, GV, MSC, RF, EV, GA, RF participated in the patients’ selection, in clinical assessment and data collection and management. ML and RR interpreted the results and drafted the manuscript. RR, AC, MSC, AM, EL, CS, AC, AS, LS, AD and RS reviewed the paper. All the authors made substantive intellectual contributions to the study, reviewed the article and gave their final approval for the version being submitted.
Funding This study was not financially supported by any pharmaceutical company.
Competing interests RR has received honoraria and speaker fees from Novartis, AbbVie, Pfizer, MSD, Janssen. AM has received honoraria and speaker fees from AbbVie, Pfizer, MSD, UCB, Novartis, Janssen, Eli-Lilly. LS speaker from Jansen, Novartis, Pfizer, UCB, MSD, Sanofi.
Patient consent for publication Not required.
Ethics approval Patients’ written consents were obtained according to the Declaration of Helsinki, when they were first entered into the database for treatment. The Ethics Committee’s approval was obtained from all participating centerscentres (approval no. 23 943), as well as the written informed consent for the anonymous use of personal data from every patient, in compliance with Italian Legislative Decree 196/2003.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Please contact the corresponding author: Roberta Ramonda (roberta.ramonda@unipd.it).