Article Text

Abstract
Objectives To evaluate the long-term safety and efficacy of belimumab in patients with systemic lupus erythematosus (SLE) from Japan and South Korea.
Methods In this phase III, open-label continuation study (BEL114333; NCT01597622), eligible completers of BEL113750 (NCT01345253) or BEL112341 (NCT01484496) received intravenous belimumab 10 mg/kg every 28 days for ≤7 years. Primary endpoint was safety. Secondary endpoints: SLE Responder Index (SRI)4 response rate, proportion of patients meeting individual SRI4 criteria, SLE flares and prednisone use. Analyses were based on observed data from the first belimumab exposure (either in parent or current study) through to study end.
Results Of 142 enrolled patients who received belimumab, 73.2% completed the study. The study population comprised patients with moderate SLE, mean (SD) Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI) baseline score of 9.3 (3.9) and 98.6% receiving corticosteroids. Most patients (97.9%) experienced adverse events (AEs); 33.8% experienced serious AEs. Increase in SRI4 (Year 1, Week 24: 47.8%; Year 6, Week 48: 68.2%) and SELENA-SLEDAI responders suggested reductions in disease activity. Proportions of patients with no worsening in Physician Global Assessment/no new organ damage remained stable throughout. Severe SLE flares occurred in 14.8% of patients. Among patients with baseline prednisone-equivalent dose >7.5 mg/day (n=81), the median (min, max) number of days anytime post-baseline that the daily dose was ≤7.5 mg/day or had been reduced by 50% from baseline was 584 (0, 2267).
Conclusions Favourable safety profile and treatment responses were maintained for ≤7 years in patients with SLE from Japan and South Korea.
- biological therapy
- B-lymphocytes
- cytokines
- lupus erythematosus
- systemic
Data availability statement
Data are available upon reasonable request. Anonymised individual patient data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Belimumab is a recombinant, humanised immunoglobulin G1 lambda (IgG1λ) monoclonal antibody that binds to and inhibits the action of soluble human B-lymphocyte stimulator (BLyS) protein.
In double-blind, placebo-controlled trials of patients with systemic lupus erythematosus (SLE), intravenous belimumab treatment reduces disease activity and new organ damage accrual and has an acceptable safety and tolerability profile.
Long-term data on the efficacy and safety of belimumab are limited in Japanese and South Korean populations.
What does this study add?
Belimumab was well tolerated and appeared to reduce disease activity and slow organ damage progression; also, it contributed to corticosteroid dose reduction over the longer term (up to 7 years) in patients with SLE in Japan and South Korea.
How might this impact on clinical practice or future developments?
The results of the presented analyses support a positive benefit–risk profile of treatment with belimumab as an add-on to standard therapy in patients with active SLE in Japan and South Korea.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a systemic autoimmune disorder characterised by loss of immune tolerance against nuclear autoantigens.1–3 Patients with SLE have elevated levels of B-lymphocyte stimulator (BLyS) protein, which promotes abnormal B-cell activation and differentiation.4–6 B cells produce autoantibodies targeting nuclear components, such as anti-double-stranded deoxyribonucleic acid (anti-dsDNA), which can lead to irreversible organ damage and early mortality.7
The prevalence and severity of SLE are greater in non-Caucasian populations (Hispanic, African descendant and Asian) than in Caucasian populations,8 suggesting a potential genetic predisposition to SLE in these groups. In Japan, SLE is estimated to affect 4.3–37.7 people per 100 000 and in South Korea the estimated prevalence is 20.6–26.5 people per 100 000.9 10 Despite the availability of various standard therapies, such as corticosteroids and immunosuppressants, many patients continue to experience a considerable disease burden.11–13 In addition, prolonged use of such therapies is often associated with frequent adverse events (AEs); thus, there remains a need for therapeutic alternatives, particularly in patients with high disease activity.14 15
Belimumab is a recombinant, humanised immunoglobulin G1 lambda (IgG1λ) monoclonal antibody (mAb) that binds to BLyS, antagonising its biological activity.16 Intravenous (IV) belimumab 10 mg/kg is approved in Europe, the USA, Japan and South Korea for the treatment of patients with active autoantibody-positive SLE receiving standard therapy.17–20 The efficacy and safety of belimumab in patients with SLE were examined in previous phase III trials, which demonstrated that belimumab-treated patients have reductions in disease activity, new organ damage accrual and corticosteroid dose.21–24 Long-term continuation studies involving 7–13 years of belimumab treatment have confirmed that efficacy and acceptable safety and tolerability profile were maintained over this time.25–27 However, limited data exist on the long-term efficacy and safety of belimumab in patients with SLE from Japan and South Korea. Therefore, the objective of this continuation study was to evaluate the long-term safety, tolerability and efficacy of belimumab in patients with SLE from Japan and South Korea.
Methods
Study design
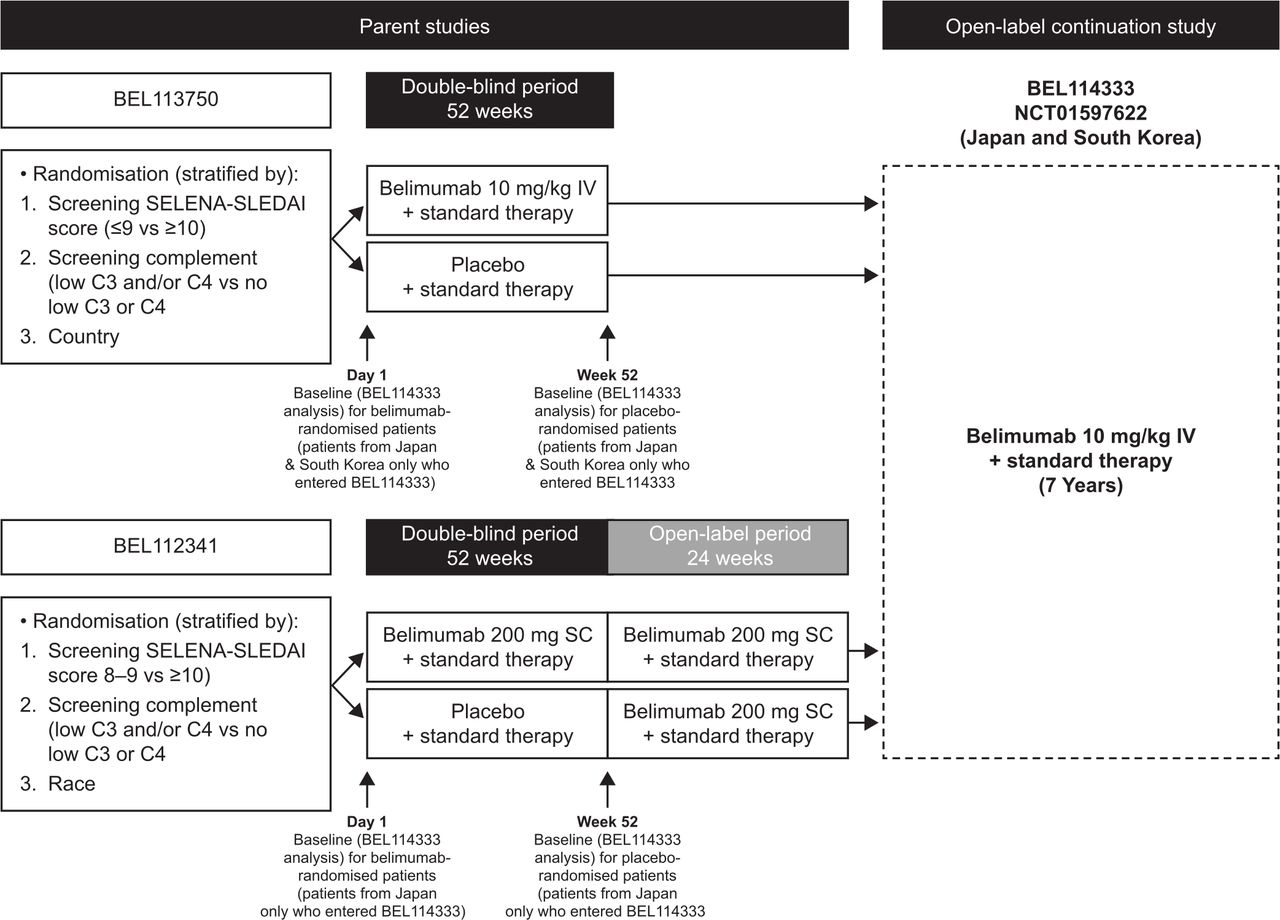
This was a phase III, open-label, multicentre, continuation study of belimumab (GlaxoSmithKline Study BEL114333; ClinicalTrials.gov identifier NCT01597622) plus standard therapy conducted at 27 study centres across Japan and South Korea in patients with SLE who completed the parent studies, the 52-week double-blind phase of the BEL113750 (NCT01345253) study receiving IV belimumab in Japan and South Korea,24 or the subcutaneous (SC) open-label phase of the BEL112341 (NCT01484496) study in Japan (figure 1).28 Eligible patients entering this study received belimumab 10 mg/kg IV infused over 1 hour every 28 days for up to 7 years plus standard therapy, irrespective of their randomised treatment in the parent studies. Patients were scheduled to receive their first belimumab dose 4 weeks (range 2–8 weeks) after their last IV dose in study BEL113750 or the first dose of IV belimumab 1 week (±1 week) after the last dose of SC belimumab in study BEL112341. Patients continued to receive belimumab unless specific stopping criteria occurred (online supplemental materials).
Supplemental material
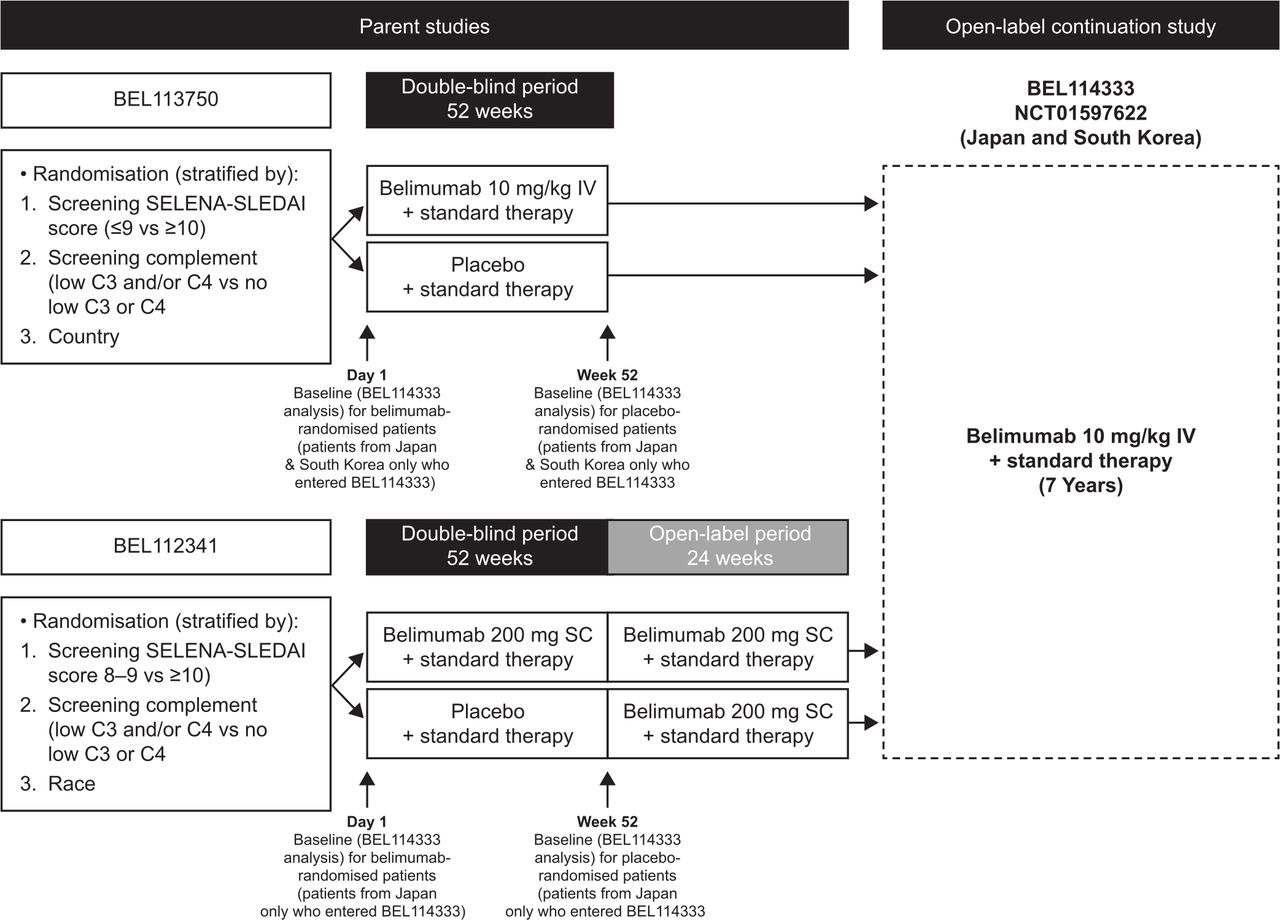
Study design. Note: Due to the additional 6-month open-label extension for SC patients entering BEL114333, there is a 6-month offset in the data capture between the parent studies. C3/C4, complement 3/4; SC, subcutaneous; SELENA-SLEDAI, Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index; SLE, systemic lupus erythematosus.
Blinding
BEL113750 study blinding was maintained by the sponsor until all double-blind data were locked, and clinical sites remained blinded to BEL113750 treatment until the double-blind results were publicly disclosed. Patients from the open-label extension of the BEL112341 study were already unblinded.
Patients
Eligible patients had completed study BEL113750 or the open-label phase of study BEL112341 in study centres in Japan or South Korea. Full eligibility criteria for the parent studies have been previously published.24 28 Patients had to be on a stable SLE treatment regimen for ≥30 days prior to Day 0 of both parent studies, consisting of any of the following (alone or in combination): prednisone-equivalent dose (0–40 mg/day when used in combination with other SLE treatment or 7.5–40 mg/day when used alone), antimalarials, non-steroidal anti-inflammatory drugs or any other immunosuppressants or immunomodulatory therapy (eg, methotrexate, azathioprine, leflunomide, mycophenolate, mizoribine, calcineurin inhibitors, sirolimus, oral cyclophosphamide, 6-mercaptopurine or thalidomide), at the discretion of the investigator (online supplemental materials).
Further key eligibility criteria and permissible and prohibited medications are listed in the online supplemental materials.
Endpoints and assessments
The primary endpoint was safety, which was assessed by the incidence of AEs, serious AEs (SAEs) and AEs of special interest (AESIs), clinical laboratory tests and immunogenicity throughout the study. Treatment-emergent AEs were defined as any AE that occurred or pre-existing event that worsened in severity or relatedness after receipt of the first belimumab dose (refer to the Statistical analysis section). AEs were assessed at each infusion visit and other safety endpoints were monitored at regular intervals, until the 16-week follow-up visit (post final belimumab dose). For further information refer to online supplemental materials.
The key secondary efficacy endpoint was observed proportion of SLE Responder Index (SRI)4 responders (defined as a ≥4-point reduction from baseline in Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI) score, no worsening in Physician Global Assessment (PGA; <0.3-point increase from baseline) and no new British Isles Lupus Assessment Group (BILAG) 1A/2B organ domain scores) assessed at scheduled 24 weeks and 4 weeks post-final belimumab dose (exit visit).
Additional secondary/other efficacy endpoints included the proportion of patients meeting each of the three SRI4 response component criteria individually; median time to first severe SELENA-SLEDAI Flare Index (SFI) flare; proportion of patients experiencing SFI flare, BILAG 1A/2B flare (≥1 new 1A or ≥2 new 2B scores) and renal flare at any time post first dose of belimumab; the cumulative number of days when prednisone dose was ≤7.5 mg/day and/or reduced by 50% from baseline and change from baseline in SELENA-SLEDAI and PGA scores. Efficacy endpoints were assessed at scheduled 24-week visits and exit visit. Change from baseline in Systemic Lupus International Collaborating Clinics (SLICC)/American College of Rheumatology (ACR) Damage Index (SDI) was assessed at Week 48 annually and at exit visit.
Change from baseline in serum IgG, anti-dsDNA autoantibodies, complement (C3, C4), B cells and B-cell subsets was also assessed. B-cell subsets were analysed centrally in patients from selected sites in Japan only.
AEs coding and the definition of SFI flares are provided in online supplemental materials.
Statistical analysis
Enrolment in this study was dependent on the number of patients in Japan and South Korea who completed the BEL113750 and BEL112341 studies; therefore, no sample size calculations were performed. Baseline was defined as the last available value prior to belimumab initiation: Day 1 of the parent study for patients randomised to belimumab and Week 52 of the parent study for patients randomised to placebo (figure 1). As such, exposure data to belimumab during parent studies were included for patients who received their first dose in the parent study and subsequently enrolled in BEL114333. Analyses were descriptive, based on observed data, and summarised relative to baseline. No follow-up data were collected post study. All data summaries and analyses were performed using SAS software, V.9.3 or higher (SAS Institute).
The safety population was defined as all patients who received ≥1 dose of belimumab during the BEL114333 study. AEs were summarised at any time post first dose of belimumab and by year of belimumab treatment, starting from first belimumab dose (baseline). All safety and efficacy endpoints were assessed in the safety population. Exit visits were slotted to the next scheduled visit for each efficacy endpoint.
Data from patients who received SC belimumab in the BEL112341 study and who received IV belimumab in the BEL113750 study were combined and analysed together. Data for the SDI, IgG and B cells and B-cell subsets were presented by parent study and by the timepoints at which they were collected, because of differences in data collection timepoints.
Patient and public involvement
This study did not involve patients or the public in the design or implementation of the study or the dissemination of its results.
Results
Patient disposition and baseline characteristics
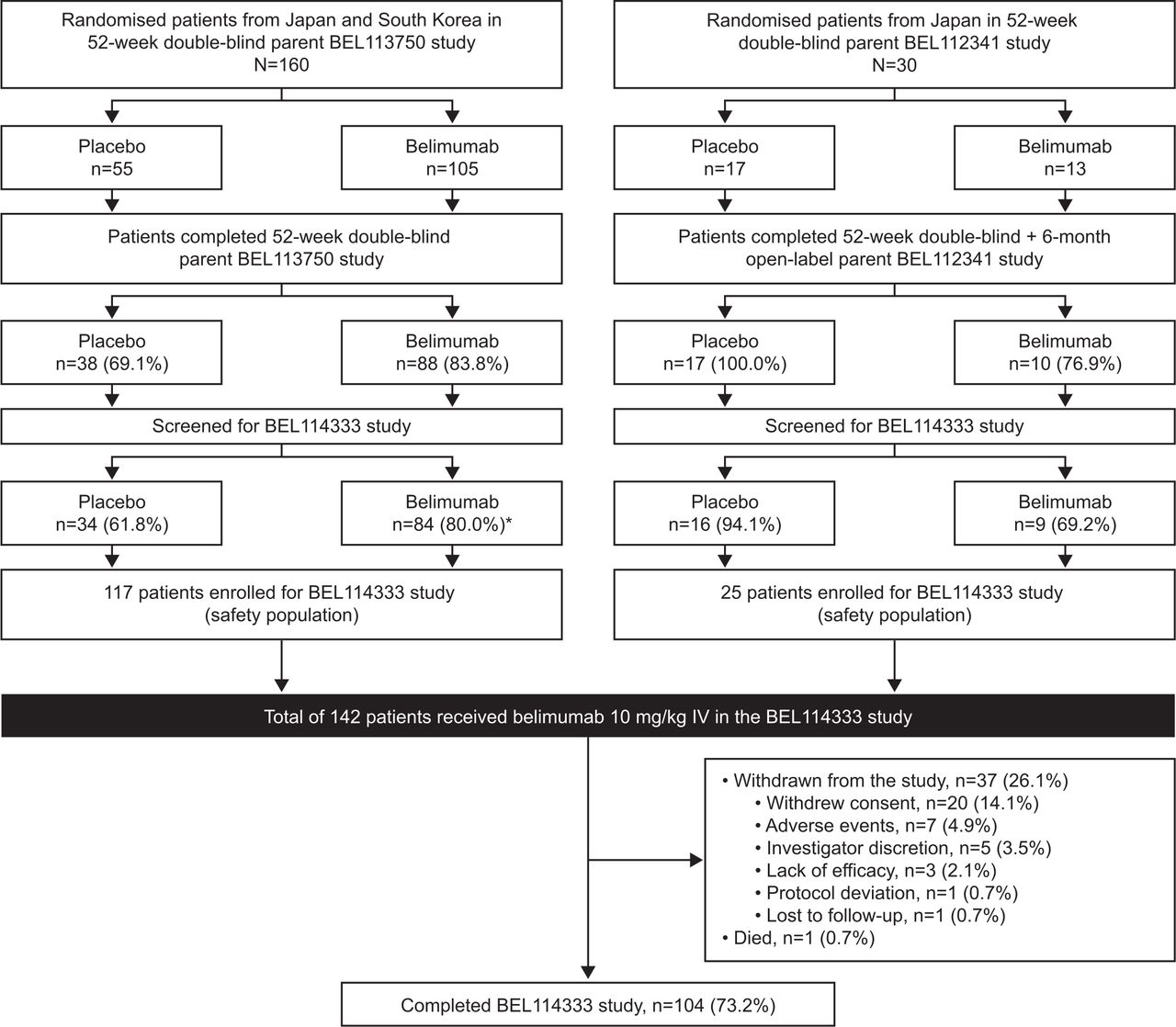
The first patient was enrolled in the continuation study (BEL114333) on 11 June 2012, and the last patient last visit for this study was on 13 September 2018. Of the 190 patients from Japan and South Korea who were randomised in the parent studies, 153 completed the respective parent studies and 142 (Japan: n=72; South Korea: n=70) enrolled in the continuation study and were included in the safety population. In total, 73.2% (104/142) of patients completed the continuation study; 1 patient died (0.7%) and 37 (26.1%) withdrew from the study, mainly because of withdrawal of consent (14.1%) and AEs (4.9%) (figure 2). The median (min, max) duration of belimumab exposure was 1171 (87, 2432) days, with a total belimumab exposure of 458.9 patient-years, including exposure during parent studies.

Patient disposition. *One screened patient in the belimumab treatment group did not enter BEL114333 due to a serious AE of chronic pancreatitis which started during the parent Study BEL113750. Note: Study completers included all patients who transferred to a different protocol or who were still participating in the study at the time of the sponsor decision to close the study when belimumab became commercially available in Japan and South Korea. There were eight adverse events (AEs) resulting in the withdrawal of seven patients from the study: spontaneous abortion, ALT increased, angiocentric lymphoma, AST increased, asphyxia, pleurisy, bacterial pneumonia and skin ulcer. ALT, alanine aminotransferase; AST, aspartate aminotransferase; IV, intravenous.
Demographic and baseline disease characteristics are presented in table 1. A total of 14.1% of patients had no BILAG domain involvement (A or B) and 51.4% of patients had a baseline SELENA-SLEDAI Score ≤9 (table 1). The mean (SD) baseline SELENA-SLEDAI Score was 9.3 (3.9). Patients who were randomised to placebo in the parent studies and subsequently entered in BEL114333, had lower mean SELENA-SLEDAI and PGA scores at baseline (prior to first dose of belimumab) compared with patients randomised to belimumab (online supplemental table 1).
Patient demographics and baseline clinical characteristics (safety population; N=142)
Most patients (98.6%) received corticosteroids at baseline, 40.8% received antimalarials and 76.1% received immunosuppressants, with tacrolimus being the most commonly received (table 1).
Concomitant SLE medications permitted during the study are shown in online supplemental table 2.
Safety
Overall, 97.9% (139/142) of patients experienced ≥1 treatment-emergent AE and most AEs were mild or moderate in severity. The most frequent AEs included nasopharyngitis (60.6%), headache (28.2%) and cough, herpes zoster and viral upper respiratory infection (each 18.3%) (table 2). AE rate for any event at any time post first dose of belimumab was 477.2 events/100 patient-years (table 3).
Overall summary of treatment-emergent AE* incidence by year interval (safety population; N=142)
Event rates of treatment-emergent AEs by year interval (safety population, N=142)
Serious treatment-emergent AEs were reported in 33.8% (48/142) of patients, with the most frequently reported SAEs by system organ class being infections and infestations. Cellulitis, contusion and herpes zoster were the most frequently reported SAEs (reported in three patients each (2.1%); table 2). The highest SAE rate by system organ class was infections and infestations (6.4 events/100 patient-years) (table 3).
Overall, 60.6% (86/142) of patients experienced a treatment-emergent AE considered by the investigator to be at least possibly treatment related (table 2). Treatment-related AEs with >5% incidence at any time post first dose of belimumab were nasopharyngitis (16.9%) and herpes zoster (9.9%; data not shown).
In total, 4.9% (7/142) of patients experienced an AE leading to treatment discontinuation/study withdrawal (1.7 events/100 patient-years). One (0.7%) death was reported during the follow-up period due to endocarditis that began 55 days after the final belimumab dose (considered by the investigator to be unrelated to study treatment) (tables 2 and 3).
AESI rates per 100 patient-years are presented in table 3. One malignancy (angiocentric lymphoma) occurred in Year 1–2; the overall rate for malignancies at any time post first dose of belimumab was 0.2 events/100 patient-years. There were no serious anaphylaxis/hypersensitivity reactions. One case of tuberculosis was reported during Year 2–3 (overall rate of 0.2 events/100 patient-years). One serious event of depression was reported (0.2 events/100 patient-years). There were no completed suicides or suicide attempts.
The annual incidence and rate of AEs of all categories, as well as AESI generally remained stable or declined throughout the study.
No trends of clinical concern were observed over time with regard to incidence of grade 3 or 4 values in haematology parameters, clinical chemistry and urinary values. There were no grade 3 or 4 serum IgG values reported post first dose of belimumab.
One patient from Japan tested positive for antidrug antibodies at the exit visit (4 weeks post last dose of belimumab), but they were confirmed as negative at the 16-week follow-up visit. This patient experienced maculopapular rash, cellulitis, thrombophlebitis and herpes zoster, which were considered by the investigator as unrelated to study treatment.
Efficacy results
Overall, the proportion of SRI4 responders during belimumab treatment increased from 47.8% (65/136) at Year 1, Week 24, to 68.2% (15/22) at Year 6, Week 48 (figure 3). Similarly, the proportion of patients with a ≥4-point reduction from baseline in SELENA-SLEDAI score increased from 51.5% (70/136) in Year 1, Week 24, to 77.3% (17/22) in Year 6, Week 48. The proportion of patients with no worsening in PGA (range of 91.3%–100.0%) and no new BILAG 1A/2B domain scores remained stable over time (range of 93.3%–100.0%).

{kind=link}
{kind=link}
{kind=link}
SRI4 response rate during belimumab treatment over time (safety population, N=142). Baseline was defined as the last available value prior to belimumab initiation: Day 1 of the parent study for patients randomised to belimumab and Week 52 of the parent study for patients randomised to placebo. SLE, systemic lupus erythematosus; SRI, SLE Responder Index.
Only 14.8% (21/142) of patients experienced ≥1 severe SFI flare post first dose of belimumab (24 severe flares; 5.2 events/100 patient-years). For patients who had a severe flare post first dose of belimumab, the median (IQR) time to first severe flare post first dose of belimumab was 585 (153.0, 882.0) days. Overall, 75.4% (107/142) of patients experienced any SFI flare post first dose of belimumab (75.8 events/100 patient-years) during belimumab treatment. The median (IQR) time to first SFI flare was 198 (89, 882) days. Only 25.4% (36/142) of patients experienced ≥1 new BILAG 1A/2B flare and 17.6% (25/142) had ≥1 new BILAG A flare post first dose of belimumab. A total of 30.3% (43/142) of patients experienced a renal flare post first dose of belimumab.
A total of 81 patients received a prednisone-equivalent dose >7.5 mg/day at baseline. The median (min, max) number of days any time post first dose of belimumab that the daily prednisone-equivalent dose was ≤7.5 mg/day or had been reduced by 50% from baseline was 584 (0, 2267). For these patients, the percentage receiving ≤7.5 mg/day prednisone dose at any visit from Year 1, Week 24, up to Year 6, Week 48, ranged from 22.2% (18/81) to 76.9% (10/13), respectively. For patients receiving ≤7.5 mg/day prednisone dose at baseline (n=61), the percentage receiving >7.5 mg/day prednisone dose at any visit from Year 1, Week 24, up to Year 6, Week 48, ranged from 8.2% (5/61) to 10.0% (1/10), respectively. For all patients, the median (IQR) per cent change from baseline in prednisone-equivalent dose decreased over time. At Year 3, Week 48 (n=90), the median (IQR) per cent change from baseline in prednisone-equivalent dose was −33.33 (−50.0, –6.7) and remained decreased through to study end.
Mean SELENA-SLEDAI Score and the mean PGA Score generally improved over time (online supplemental figure 1). There was little change in SDI Score from baseline. No more than four patients from BEL113750 and no more than two patients from BEL112341 had SDI worsening (change >0) compared with baseline at any Week 48 and Week 24 visit, respectively.
Biomarkers
The IgG median per cent change from baseline decreased only slightly over time (online supplemental figure 2A). The median anti-dsDNA per cent change from baseline also decreased (online supplemental figure 2B). The median per cent change in C3 and C4 levels increased and generally remained at a raised level above baseline during belimumab treatment (online supplemental figure 2C). The levels of B-cell subsets either decreased or remained stable throughout the study (online supplemental table 3).
Discussion
The results of this long-term, open-label continuation study of patients with SLE from Japan and South Korea demonstrated high retention of patients, with 104/142 patients completing the study, which reflects the effectiveness and safety of intravenous belimumab as an add-on to standard therapy for up to 7 years. The safety profile observed is consistent with that seen in previous randomised controlled trials and other continuation studies of 7–13 years’ duration.21 22 24–27 29 The incidence of treatment-emergent AEs, including AESIs, generally remained stable or declined over time, and the overall immunogenicity response rate was low. The 0.7% incidence of mortality in the current study is similar to the 0.7%–1.5% incidence reported in the previous 7–8-year belimumab continuation studies.25 26 30
In this long-term continuation study, the treatment benefits of belimumab in patients with SLE from Japan and South Korea were maintained over time (up to 7 years). Of the patients remaining in the study at Year 6, Week 48, over two-thirds were SRI4 responders.
Although some patients have received hydroxychloroquine, an antimalarial agent recommended for patients with SLE for minimising risk of flares, at the discretion of the investigators, the number of patients receiving it in this study was low as the study began before the hydroxychloroquine was approved in Japan.31 Nonetheless, the majority of patients did not experience severe flares, consistent with belimumab use as observed in other long-term continuation studies.25 27
The results of this study suggest that belimumab allows for corticosteroid dose reduction, as is evident from the progressive decrease in median (IQR) percentage change from baseline in prednisone-equivalent dose over time. These findings support the results of BLISS-52 and BLISS-76 studies, in which belimumab therapy showed a reduction in prednisone dose compared with standard therapy alone.21 22
These benefits were coupled with increasing improvements over time in anti-dsDNA, C3 and C4, which is consistent with the results from previous studies and with the mechanism of action of belimumab as an antibody that specifically targets and inhibits the activity of BLyS.21 22 Together these outcomes suggest that decreases in disease activity with belimumab were associated with limited new organ damage accrual.
Limitations of interpretation of the results include the small sample size, particularly at later study visits, and the absence of a placebo control group meant that treatment comparisons could not be made. However, the number of Japanese and Korean patients enrolled in this open-label study (N=142) was proportionate to the number of Japanese and Korean patients randomised in the double-blind phase of the parent studies (BEL113750, N=160; BEL112341, N=30). The inclusion of a placebo comparator group in a long-term, 7-year study would not be feasible and would also be considered unethical. Additionally, as patients had to complete the parent studies, the sample of patients included in this continuation study was self-selected and potentially more likely to respond well to belimumab over time. Furthermore, the results of this study are based on observed data with no imputation analyses for withdrawal, which may also bias the findings. Patients randomised to placebo in the double-blind period of the parent studies had lower mean SELENA-SLEDAI and PGA scores at baseline than patients randomised to belimumab, which could be due to different baseline timepoints (baseline for the belimumab group was at the start of the double-blind parent study; baseline for the placebo group was at the start of open-label period). This phenomenon was observed also in a previous belimumab study of similar design.32 Placebo-treated patients had already completed the parent studies during which their standard therapy and care would have been optimised and their disease activity would have been likely to decrease. Despite these limitations, the results of this open-label continuation analysis are consistent with the known safety and efficacy profile of belimumab.21 22 24 25 27 33
In conclusion, the safety results of this 7-year study of patients with SLE from Japan and South Korea are consistent with those observed in previous 7–13-year open-label continuation studies of belimumab added to standard therapy. Reductions in disease activity and corticosteroids dose, in addition to a low rate of new organ damage accrual, suggest that patients with SLE from Japan and South Korea achieve long-term benefits from belimumab treatment.
Data availability statement
Data are available upon reasonable request. Anonymised individual patient data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by local institutional review boards and/or ethics committees and conducted in accordance with the International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Good Clinical Practice ethical principles and the Declaration of Helsinki. All patients provided written informed consent.
Acknowledgments
The authors would like to thank the participating patients and their families, clinicians and study investigators.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published online. In paragraph 4 under the safety section, the overall proportion of patients experiencing treatment-emergent adverse events that the investigator considered to be at least possibly related to belimumab treatment was corrected from 57.0% (81/142) to 60.6% (86/142).2. In the same paragraph, for the treatment-related adverse events with >5% incidence at any time post first dose of belimumab, the proportion of patients experiencing nasopharyngitis was changed from 15.5% to 16.9% and herpes zoster from 8.5% to 9.9%.3. Some safety data for treatment-related adverse events in Tables 2 and 3 have been updated.
Contributors Conception or design: DB, PC, BJ, DAR and MC. Acquisition of data: YT and S-CB. Data analysis or interpretation: all authors.
Funding This study was funded by GlaxoSmithKline (GSK Study BEL114333; NCT01597622). Editorial support was provided by Olga Conn, PhD, of Fishawack Indicia Ltd., UK, part of Fishawack Health, and was funded by GSK.
Competing interests YT received research grants from Mitsubishi-Tanabe, Chugai, AbbVie, Takeda, UCB, Daiichi-Sankyo and Eisai and speaking fees and/or honoraria from Daiichi-Sankyo, Eli Lilly, Novartis, YL Biologics, Bristol-Myers, Eisai, Chugai, AbbVie, Astellas, Pfizer, Sanofi, Asahi-Kasei, GlaxoSmithKline, Mitsubishi-Tanabe, Gilead and Janssen. S-CB has no conflicts of interest to declare. DB, PC, MC, KD, BJ, RK, JL, PM and DAR are employees of GlaxoSmithKline and hold stocks and shares in the company.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note MC and RK were employees of GlaxoSmithKline at the time of the study.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.