Article Text

Abstract
Objectives To report the efficacy, safety and patient-reported outcome measures (PROs) of tofacitinib modified-release 11 mg once daily plus methotrexate in patients with rheumatoid arthritis (RA) from the open-label phase of Oral Rheumatoid Arthritis Trial (ORAL) Shift.
Methods ORAL Shift was a global, 48-week, phase 3b/4 withdrawal study in patients with moderate to severe RA and an inadequate response to methotrexate. Patients received open-label tofacitinib modified-release 11 mg once daily plus methotrexate; those who achieved low disease activity (LDA; Clinical Disease Activity Index (CDAI)≤10) at week 24 were randomised to receive blinded tofacitinib 11 mg once daily plus placebo (ie, blinded methotrexate withdrawal) or continue with blinded tofacitinib 11 mg once daily plus methotrexate for another 24 weeks. Efficacy, PROs and safety from the open-label phase are reported descriptively.
Results Following screening, 694 patients were enrolled and received tofacitinib plus methotrexate in the open-label phase. At week 24, 527 (84.5%) patients achieved CDAI-defined LDA. Improvements from baseline to weeks 12 and 24 were generally observed for all efficacy outcomes (including measures of disease activity, and response, LDA and remission rates) and PROs. Adverse events (AEs), serious AEs and discontinuations due to AEs were reported by 362 (52.2%), 20 (2.9%) and 41 (5.9%) patients, respectively. No deaths were reported.
Conclusions Tofacitinib modified-release 11 mg once daily plus methotrexate conferred improvements in disease activity measures, functional outcomes and PROs, with most (84.5%) patients achieving CDAI-defined LDA after 24 weeks of open-label treatment; the safety profile was generally consistent with the historic safety profile of tofacitinib.
Funded by Pfizer Inc; NCT02831855.
- arthritis
- rheumatoid
- Methotrexate
- patient reported outcome measures
Data availability statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
ORAL Shift was the first study of tofacitinib modified-release 11 mg once daily administered in combination with methotrexate in a global population of patients with rheumatoid arthritis and an inadequate response to methotrexate.
Results from the 24-week double-blind phase of ORAL Shift showed that withdrawal of methotrexate may be considered in patients with rheumatoid arthritis who achieve low disease activity (LDA) with tofacitinib 11 mg once daily plus methotrexate.
What does this study add?
In the 24-week open-label phase of ORAL Shift, clinically meaningful efficacy was observed in patients with rheumatoid arthritis receiving tofacitinib modified-release 11 mg once daily plus methotrexate for all endpoints, including improvements in disease activity measures, functional outcomes and patient-reported outcomes. In addition, the safety profile of tofacitinib modified-release 11 mg once daily plus methotrexate was generally consistent with the historic safety profile of tofacitinib immediate-release 5 mg twice daily.
In post hoc analyses, lower baseline disease activity (Clinical Disease Activity Index score) was a significant predictor of LDA or remission (defined by multiple outcomes) following 24 weeks of open-label treatment with tofacitinib 11 mg once daily plus methotrexate.
Key messages
How might this impact on clinical practice or further developments?
Considering the efficacy and safety profiles demonstrated here for tofacitinib modified-release 11 mg once daily plus methotrexate and previously for tofacitinib immediate-release 5 mg twice daily plus methotrexate in a similar patient population, there are options available in the treatment landscape to accommodate patient preferences, which may improve treatment adherence and, ultimately, patient outcomes.
Introduction
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. The efficacy and safety of tofacitinib immediate-release 5 and 10 mg twice daily, administered as monotherapy or in combination with conventional synthetic disease-modifying antirheumatic drugs (DMARDs; predominantly methotrexate) in patients with moderate to severe rheumatoid arthritis, have been demonstrated in phase 31–7 and phase 3b/48 randomised controlled trials of up to 24 months' duration, and in long-term extension studies up to 114 months' observation.9–11
Different treatment options may accommodate patient preferences, improve treatment adherence and, ultimately, improve patient outcomes.12 To provide a once-daily dosing option, a modified-release formulation of tofacitinib was developed. The tofacitinib immediate-release 5 mg twice-daily and modified-release 11 mg once-daily formulations have comparable pharmacokinetic parameters at steady state,13 and an analysis of the US Corrona rheumatoid arthritis registry demonstrated comparative effectiveness of these formulations in real-world patients.14 Furthermore, clinically meaningful improvements in rheumatoid arthritis were observed with both formulations in a randomised controlled trial of Japanese patients, although non-inferiority of tofacitinib modified-release 11 mg once daily versus tofacitinib immediate-release 5 mg twice daily (primary efficacy endpoint) was not demonstrated.15 The double-blind, double-dummy design of this trial required patients who received the modified-release formulation of tofacitinib to take a pill twice daily,15 limiting measurement of potential real-world benefits of the modified-release formulation in a once-daily regimen.
Oral Rheumatoid Arthritis Trial (ORAL) Shift is the first study of tofacitinib modified-release 11 mg once daily administered in combination with methotrexate in a global population of patients with rheumatoid arthritis. Results of the 24-week, double-blind methotrexate withdrawal phase of ORAL Shift were reported previously,16 and showed that non-inferiority of tofacitinib modified-release 11 mg once-daily monotherapy versus tofacitinib modified-release 11 mg once daily plus methotrexate (primary efficacy endpoint) was met, with respect to change in disease activity (defined as Disease Activity Score in 28 joints, erythrocyte sedimentation rate (DAS28-4(ESR))). Specifically, patients who achieved low disease activity (LDA; Clinical Disease Activity Index (CDAI) ≤10) with tofacitinib modified-release 11 mg once daily plus methotrexate, and subsequently withdrew methotrexate, did not have significant worsening of disease activity or unexpected safety issues. Overall, results indicated that in patients who achieve LDA with tofacitinib modified-release 11 mg once daily plus methotrexate, withdrawal of methotrexate may be considered.16
We report results from the 24-week open-label phase of ORAL Shift, which evaluated the efficacy, patient-reported outcomes (PROs) and safety of tofacitinib modified-release 11 mg once daily plus methotrexate in patients with moderate to severe rheumatoid arthritis and an inadequate response to methotrexate. Post hoc analyses, which assessed baseline predictors of LDA and remission after 24 weeks of tofacitinib modified-release 11 mg once daily plus methotrexate treatment, are also presented.
Methods
Study design
ORAL Shift was a 48-week, phase 3b/4 withdrawal study in patients with moderate to severe rheumatoid arthritis and an inadequate response to methotrexate. The study included a 30-day screening phase, a 24-week open-label run-in phase and a 24-week randomised, double-blind, placebo-controlled, non-inferiority methotrexate withdrawal phase (online supplemental figure 1). Baseline was defined as day 1 of the open-label phase.
Supplemental material
The study involved 109 centres across 16 countries.
Patients
Full patient inclusion and exclusion criteria have been reported previously.16 Patients eligible to enter the study were aged ≥18 years, met the 2010 American College of Rheumatology (ACR) and European Alliance of Associations for Rheumatology (EULAR) Classification Criteria for rheumatoid arthritis17 before/at the screening visit, had ≥4 tender/painful joints on motion and ≥4 swollen joints (based on 28 joint counts) at screening and baseline, and moderate to severe rheumatoid arthritis (moderate to high disease activity defined as CDAI >10 and DAS28-4(ESR) ≥3.2) at baseline. Eligible patients must have received oral methotrexate continuously for ≥4 months before screening at a stable weekly dose. They must have had an inadequate response to methotrexate, per residual disease activity that met study entry criteria.
Key exclusion criteria included history of an insufficient response to ≥2 biological (b)DMARDs or prior treatment with tofacitinib. Following a protocol amendment after initiation of the study, patients previously treated with baricitinib or any other investigational Janus kinase inhibitors were also ineligible.16 Patients were also excluded if they received live or live attenuated vaccines (≤4 weeks before the first dose of study drug or planned ≤6 weeks after study drug discontinuation) or certain prohibited concomitant medications, including bDMARDs or non-bDMARDs (other than methotrexate) within their specified washout period (≥4 weeks or five half-lives, whichever was longer, before the first dose of study drug), and corticosteroids other than stable, low-dose oral corticosteroids (doses equivalent to ≤10 mg prednisone per day for ≤4 weeks before the initial dose of study drug). Additional exclusion criteria applied to patients with: any infection requiring hospitalisation ≤6 months before the study; current, or a history of prior recurrent or disseminated herpes zoster, or active or prior active or latent tuberculosis that was inadequately treated; malignancy or history of malignancy, with the exception of adequately treated or excised non-metastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ; and any uncontrolled clinically significant laboratory abnormality.
At the end of the open-label phase, only patients who achieved LDA (CDAI ≤10) were randomised into the double-blind phase. Patients who did not achieve LDA at week 24 were discontinued from the study.
All patients provided written informed consent.
Treatment
Eligible patients were assigned to open-label treatment and received tofacitinib modified-release 11 mg once daily with methotrexate (hereafter, tofacitinib plus methotrexate) at the previously stabilised methotrexate dose for 24 weeks during this run-in phase. The sponsor provided oral tofacitinib modified-release 11 mg once-daily tablets and methotrexate 2.5 mg capsules to patients for self-administration. The methotrexate dose must have been the same as the prestudy dose and between 15 and 25 mg/week; doses between 10 and 15 mg/week were only allowed with evidence of intolerance to or toxicity from higher doses, and methotrexate doses <10 or >25 mg/week were prohibited. Patients also received supplemental oral folic acid (≥5 mg/week) or folinic acid (≥2.5 mg/week) according to local methotrexate guidelines. Details of the randomisation and masking procedures for the double-blind phase were reported previously.16
Outcomes
All primary and secondary endpoints were assessed in the double-blind phase, as reported previously.16
Efficacy endpoints assessed in the open-label phase included: mean change from baseline to weeks 12 and 24 in DAS28-4(ESR), DAS28-4 in 28 joints, C-reactive protein (DAS28-4(CRP)), CDAI, Simplified Disease Activity Index (SDAI), tender joint count (TJC; 28 joints), swollen joint count (SJC; 28 joints), Physician Global Assessment (PGA) Visual Analogue Scale (VAS) and CRP; the proportions of patients achieving LDA (DAS28-4(ESR) ≤3.2, DAS28-4(CRP) ≤3.2, CDAI ≤10, SDAI ≤11) and remission (ACR-EULAR Boolean remission criteria,18 DAS28-4(ESR) <2.6, DAS28-4(CRP) <2.6, CDAI ≤2.8 and SDAI ≤3.3) at weeks 12 and 24; the proportions of patients achieving an ACR response of at least 20% (ACR20), 50% (ACR50) and 70% (ACR70) at weeks 12 and 24; and the proportions of patients achieving a Health Assessment Questionnaire-Disability index (HAQ-DI) response (decrease from baseline of ≥0.22).
Additional endpoints included changes from baseline to weeks 12 and 24 in the following PROs: HAQ-DI, Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), Patient Assessment of Arthritis Pain (Pain VAS), Patient Global Assessment of Disease Activity (PtGA) VAS, Short Form-36 Health Survey (version 2; acute) Physical Component Summary (PCS) and Mental Component Summary (MCS) scores, eight norm-based domain scores (physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health) and the EuroQoL-Five Dimensions Health Questionnaire (EQ-5D; dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression).
Post hoc analyses assessed endpoints including: the proportions of patients achieving LDA (CDAI ≤10) by geographical region at week 24; the proportions of patients achieving LDA (DAS28-4(ESR) ≤3.2, CDAI ≤10), remission (DAS28-4(ESR) <2.6, CDAI ≤2.8) and ACR20/50/70, or HAQ-DI (decrease from baseline of ≥0.22) responses by prior bDMARD/targeted synthetic (ts)DMARD use (no prior use vs prior use and discontinuation due to inadequate pain relief, or no prior use vs prior use and discontinuation due to adverse events or other reasons) at week 24; and baseline predictors of LDA (DAS28-4(ESR) ≤3.2, CDAI ≤10) and remission (ACR-EULAR Boolean remission criteria,18 DAS28-4(ESR) <2.6, CDAI ≤2.8) at week 24.
Safety endpoints included all adverse events, including serious adverse events, discontinuations due to adverse events, adverse events of special interest (serious infections, herpes zoster, malignancies, major adverse cardiovascular events (MACE), opportunistic infections, tuberculosis, interstitial lung disease, gastrointestinal perforation or obstruction, pulmonary embolism and deep vein thrombosis) and deaths, and clinically significant abnormal laboratory parameters. An external data monitoring committee was responsible for ongoing monitoring of patient safety. External adjudication committees were established to standardise the assessment of selected safety events, including cardiovascular events, malignancies, opportunistic infections, hepatic events and gastrointestinal perforations. An internal adjudication committee was established to review and categorise interstitial lung disease events.
Statistical analysis
Full details of protocol amendments and statistical analyses for the double-blind phase (including primary and secondary endpoints) have been described previously.16
Briefly, with a two-sided type I error of 5% and an assumed SD of 1.4 for the difference between the tofacitinib monotherapy group and the tofacitinib plus methotrexate group in DAS28-4(ESR) change from randomisation, it was established that a sample size of approximately 232 patients randomised at week 24 would yield 90% power to declare non-inferiority of tofacitinib monotherapy relative to tofacitinib plus methotrexate, with a non-inferiority margin of 0.6 (as reported previously).16 Protocol amendments were subsequently made to account for the rates of enrolment, achievement of LDA and open-label phase discontinuation, and it was determined that 680 patients were required for enrolment into the open-label phase to ensure that ≥232 patients would be randomised at week 24 in the double-blind phase.
Efficacy outcomes and PROs were assessed using the full analysis set of the open-label phase, defined as patients who received ≥1 dose of tofacitinib modified-release 11 mg once daily plus methotrexate during the open-label phase. Safety was assessed in the safety analysis set, which was identical to the full analysis set.
Efficacy outcomes and PROs were summarised descriptively in the open-label phase as observed cases. Binary endpoints (ie, ACR20/50/70 and HAQ-DI response rates, and LDA and remission) were also analysed using non-responder imputation (data not shown).
Post hoc analyses of binary endpoints (ie, LDA by geographical region, and LDA, remission and ACR20/50/70 or HAQ-DI response rates by prior bDMARD/tsDMARD use) were also summarised descriptively by observed cases and using non-responder imputation (data not shown).
Post hoc analyses of baseline predictors of LDA (DAS28-4(ESR) ≤3.2, CDAI ≤10) and remission (ACR-EULAR Boolean remission criteria,18 DAS28-4(ESR) <2.6, CDAI ≤2.8) at week 24 were conducted. Patient demographics and baseline characteristics (covariates) were assessed for their significance in predicting LDA and remission in logistic regression analyses of each outcome. Covariates with p values <0.10 in the simple logistic regression models (data not shown) were included for consideration in multiple logistic regression analyses. The resulting model for each LDA and remission outcome included all selected covariates, using a stepwise procedure with an entry-criterion p value of 0.15 and a stay-criterion p value of 0.05. These final multiple logistic regression models and estimates of ORs for response and 95% CIs, are presented. The full list of covariates considered for this analysis is provided in the online supplemental file.
Safety and laboratory data for the open-label phase were summarised descriptively.
Statistical analyses were performed using SAS, V.9.4.
This trial is registered with ClinicalTrials.gov (NCT02831855), and is complete.
Results
Between 1 September 2016 and 1 November 2017, 694 patients were enrolled in the open-label phase and received tofacitinib plus methotrexate (online supplemental figure 2). Of these patients, 71 (10.2%) discontinued before the week 24 visit (end of open-label phase), including 39 (5.6%) due to adverse events.
The majority of patients were female (532 (76.7%) of 694 patients), white (594 (85.6%) patients), with a mean age of 56.8 years, mean rheumatoid arthritis disease duration of 8.8 years, and had high disease activity based on DAS28-4(ESR) >5.1 and CDAI >22 (554 (79.8%) and 583 (84.0%) patients, respectively; table 1). Half (355 (51.2%) patients) of the study population were enrolled at sites in Europe, with the majority (328 (92.4%)) of these patients from Eastern Europe. Mean baseline methotrexate dose was 16.7 mg/week and 260 (37.5%) patients were receiving concomitant oral corticosteroids at a mean dose of 5.7 mg/day.
Demographics and baseline disease characteristics of patients treated in the open-label phase
Improvements from baseline in disease activity, including DAS28-4(ESR), DAS28-4(CRP), SDAI and CDAI, were observed at weeks 12 and 24 in patients who received tofacitinib plus methotrexate during the open-label phase (table 2). Similarly, improvements from baseline in physician-reported measures, including TJC, SJC, PGA VAS and CRP were observed at weeks 12 and 24 (table 2). These outcomes also showed an improvement from week 12 to week 24.
Mean (SD) change from baseline* in efficacy outcomes and patient-reported outcomes with tofacitinib modified-release 11 mg once daily plus methotrexate at weeks 12 and 24 of the open-label phase
Furthermore, the proportion of patients achieving LDA, remission and response (ACR20/50/70 and HAQ-DI) increased from week 12 to week 24 (figure 1).
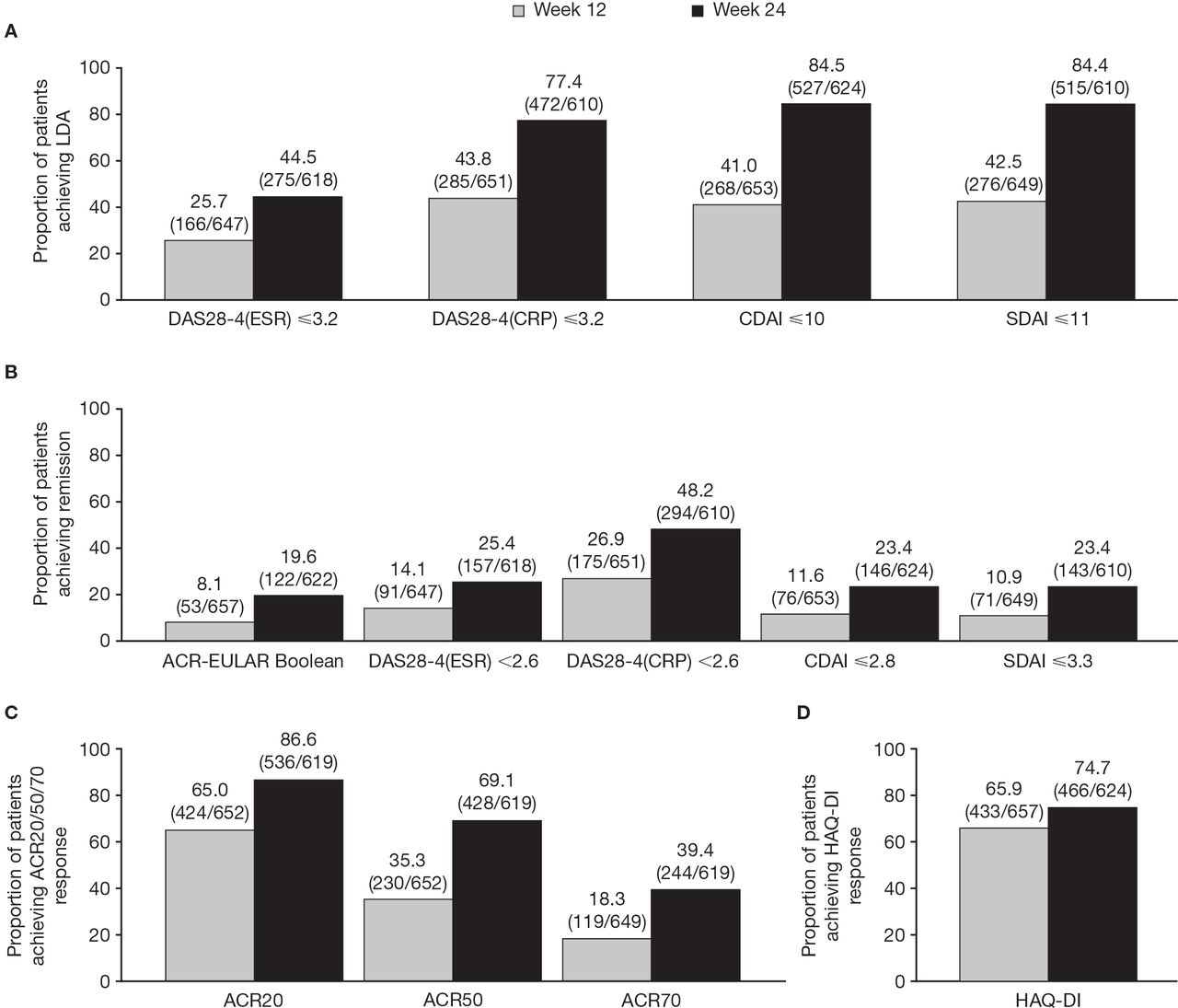
Rates of (A) LDA, (B) remission and (C) ACR20/50/70,* and (D) HAQ-DI† response rates, with tofacitinib modified-release 11 mg once daily plus methotrexate at weeks 12 and 24 of the open-label phase. Data are % (n/N). *20% (ACR20), 50% (ACR50) and 70% (ACR70) improvement from baseline in tender and swollen joint counts and 20%, 50% and 70% improvement in at least three of the five other criteria (PtGA VAS, PGA VAS, HAQ-DI, Pain VAS and CRP). †HAQ-DI decrease of at least 0.22 from baseline. Based on observed case data only. ACR, American College of Rheumatology; CDAI, Clinical Disease Activity Index; DAS28-4(CRP), Disease Activity Score in 28 joints, C-reactive protein; ESR, erythrocyte sedimentation rate; EULAR, European Alliance of Associations for Rheumatology; HAQ-DI, Health Assessment Questionnaire-Disability Index; LDA, low disease activity; n, number of patients achieving a response; N, number of patients evaluable for each outcome measure; PGA, Physician Global Assessment; PtGA, Patient Global Assessment of Disease Activity; SDAI, Simplified Disease Activity Index; VAS, Visual Analogue Scale.
Consistent with the improvements in disease activity and physician-reported measures, improvements from baseline in PROs, including HAQ-DI, FACIT-F, Pain VAS, PtGA VAS, SF-36v2 (MCS, PCS and domain scores) and EQ-5D, were observed at weeks 12 and 24 (table 2; online supplemental table 1). These outcomes also showed an improvement or remained the same (EQ-5D) from week 12 to week 24.
At week 24, 527 (84.5%) of 624 patients achieved LDA (CDAI ≤10; figure 1). In post hoc analyses of patients stratified by geographical region, the proportion who achieved CDAI-defined LDA at week 24 was numerically lower in the US (158 (68.7%) of 230 patients) versus other regions (all >90%) (online supplemental figure 3).
In post hoc analyses, when patients were stratified based on whether they had received prior bDMARDs (n=268) or tsDMARDs (n=5), there were no clinically meaningful differences in the proportions of patients achieving: LDA (DAS28-4(ESR) ≤3.2, CDAI ≤10), remission (DAS28-4(ESR) <2.6, CDAI ≤2.8), and ACR20/50/70 or HAQ-DI responses at week 24 (online supplemental table 2).
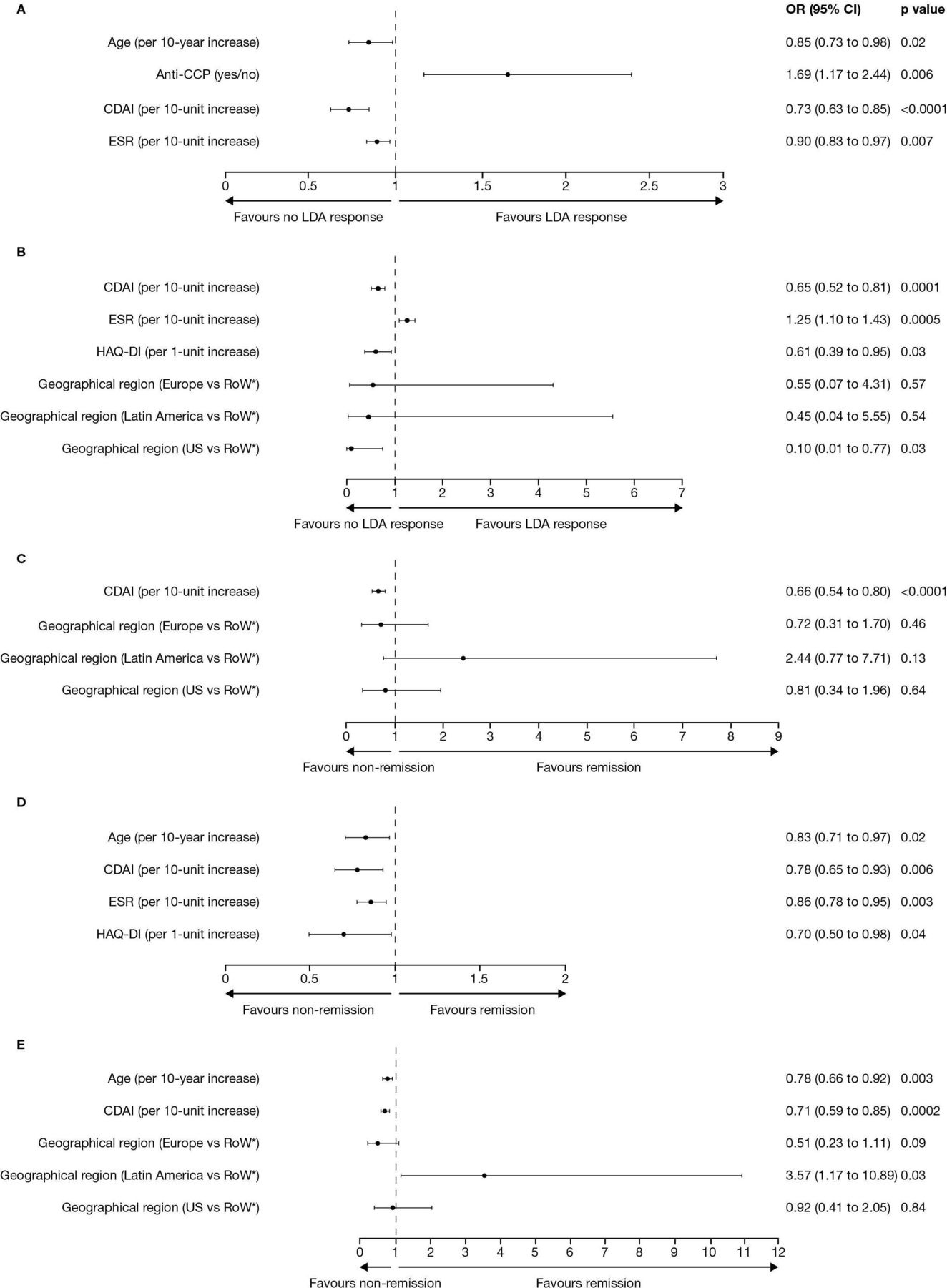
For post hoc analyses of the baseline predictors of LDA and remission with tofacitinib plus methotrexate at week 24, results from the multiple logistic regression analyses are presented in figure 2.

{kind=link}
{kind=link}
Multiple logistic regression analyses of baseline predictors of LDA, based on (A) DAS28-4(ESR) ≤3.2 and (B) CDAI ≤10, or remission, based on (C) ACR-EULAR Boolean remission criteria, (D) DAS28-4(ESR) <2.6 and (E) CDAI ≤2.8, with tofacitinib modified-release 11 mg once daily plus methotrexate at week 24 of the open-label phase. *Australia, Philippines, South Korea and South Africa. ACR, American College of Rheumatology; CCP, cyclic citrullinated peptide; CDAI, Clinical Disease Activity Index; DAS28-4(ESR), Disease Activity Score in 28 joints, erythrocyte sedimentation rate; EULAR, European Alliance of Associations for Rheumatology; HAQ-DI, Health Assessment Questionnaire-Disability Index; LDA, low disease activity; RoW, Rest of the World.
Age, anticyclic citrullinated peptide (anti-CCP) antibody test result, and baseline CDAI and ESR, were significant predictors of achieving DAS28-4(ESR)-defined LDA at week 24 (all p<0.05). Every 10-year increase in age reduced the odds of achieving LDA by 15%, and every 10-unit increase in baseline ESR and CDAI scores reduced the odds of achieving LDA by 10% and 27%, respectively. A positive anti-CCP antibody test result increased the odds of achieving LDA by 69%. Significant predictors of achieving CDAI-defined LDA at week 24 were baseline CDAI, ESR and HAQ-DI scores, as well as geographical region (all p<0.05). Every 10-unit increase in baseline CDAI score and 1-unit increase in HAQ-DI score reduced the odds of achieving LDA by 35% and 39%, respectively, whereas every 10-unit increase in baseline ESR score increased the odds of achieving LDA by 25%. Compared with the Rest of the World (Australia, Philippines, South Africa and South Korea), patients in the US were 90% less likely to achieve LDA. Overall, baseline CDAI and ESR scores were significant predictors of both LDA outcomes.
Baseline CDAI score was a significant predictor of achieving ACR-EULAR Boolean-defined remission18 at week 24 (p<0.0001); every 10-unit increase in baseline CDAI score reduced the odds of achieving remission by 34%. Significant predictors of achieving DAS28-4(ESR)-defined remission at week 24 were age and baseline CDAI, ESR and HAQ-DI scores (all p<0.05). Every 10-year increase in age reduced the odds of achieving remission by 17%, whereas every 10-unit increase in baseline ESR and CDAI scores and every 1-unit increase in HAQ-DI score reduced the odds of achieving remission by 14%, 22% and 30%, respectively. Furthermore, age, baseline CDAI score and geographical region were significant predictors of achieving CDAI-defined remission at week 24 (all p<0.05). Every 10-year increase in age and every 10-unit increase in baseline CDAI score reduced the odds of achieving remission by 22% and 29%, respectively. Patients from Latin America were 3.6 times as likely to achieve remission compared with the Rest of the World. Overall, baseline CDAI score was a significant predictor of all remission outcomes.
The incidence of all-cause adverse events, serious adverse events, severe adverse events and discontinuations due to adverse events was reported by 362 (52.2%), 20 (2.9%), 25 (3.6%) and 41 (5.9%) of 694 patients receiving tofacitinib plus methotrexate in the open-label phase, respectively (table 3). No deaths were reported in the open-label phase. No new or unexpected safety events were observed.
Safety with tofacitinib modified-release 11 mg once daily plus methotrexate during the open-label phase*
Most adverse events were mild or moderate in severity. The most common adverse events by preferred term (occurring in ≥2% of patients) were nasopharyngitis and upper respiratory tract infection.
Adverse events of special interest occurred in <1% of patients (table 3).
Liver function tests with increases ≥3 × above the upper limit of normal in aspartate aminotransferase and alanine aminotransferase occurred in 10 (1.5%) and 19 (2.8%) of 688 patients, respectively, receiving tofacitinib plus methotrexate (online supplemental table 3).
Discussion
ORAL Shift demonstrated the efficacy and safety of tofacitinib modified-release 11 mg once daily plus methotrexate for the first time in a global population of patients with rheumatoid arthritis.
In the 24-week open-label phase, clinically meaningful improvements from baseline for all disease activity and patient- and physician-reported outcomes were generally sustained at week 12 through to week 24 with tofacitinib modified-release 11 mg once daily plus methotrexate. Similarly, LDA, remission and response (ACR20/50/70 and HAQ-DI) rates were increased from week 12 to week 24, and 84.5% of patients receiving tofacitinib modified-release 11 mg once daily plus methotrexate achieved CDAI-defined LDA at week 24. Adverse events, serious adverse events and discontinuations due to adverse events were reported by 52.2%, 2.9% and 5.9% of patients, respectively. Adverse events of special interest were infrequent, occurring in <1% of patients (serious infections, herpes zoster (all non-serious), malignancies and interstitial lung disease), or in no patients (MACE, opportunistic infections, tuberculosis, gastrointestinal perforation or obstruction, pulmonary embolism/deep vein thrombosis and deaths) in the open-label phase. In all, the safety profile of tofacitinib modified-release 11 mg once daily plus methotrexate was generally consistent with the historic safety profile of tofacitinib immediate-release 5 mg twice daily.
In post hoc analyses of the open-label phase, no clinically meaningful differences were observed among the proportions of patients achieving LDA, remission or response based on their prior bDMARD/tsDMARD experience.
Achievement of LDA, or ideally remission, is the goal of the treat-to-target strategy for rheumatoid arthritis.19 20 Understanding which patients are more likely to respond to treatment will inform treatment decisions and lead to better management of patients. Baseline CDAI score was a significant predictor of all LDA and remission outcomes after 24 weeks of tofacitinib modified-release 11 mg once daily plus methotrexate. This corresponds with a similar population of patients with rheumatoid arthritis and inadequate response to methotrexate in which baseline disease activity was a significant predictor of LDA after 24 weeks of tofacitinib immediate-release 5 mg twice daily plus methotrexate.21 Of interest, younger age was a significant predictor of DAS28-4(ESR)-defined LDA and DAS28-4(ESR)-defined and CDAI-defined remission; and reduced disability per HAQ-DI was a significant predictor of CDAI-defined LDA and DAS28-4(ESR)-defined remission. Further, anti-CCP positivity was a significant predictor of DAS28-4(ESR)-defined LDA, but no other LDA or remission outcomes. Notably, disease duration, prior bDMARD and steroid use, and baseline body mass index and rheumatoid factor test results were not significant predictors of LDA or remission. Baseline CDAI score being a significant predictor of LDA and remission is not unexpected, as these endpoints are more difficult to achieve in patients with higher baseline disease activity.
Predictors of LDA and remission have been examined in other rheumatoid arthritis studies with varying results, due in part to the treatment administered and the LDA and remission criteria used. Indeed, a systematic review comprised mostly of prospective cohort studies, in which patients were treated with non-bDMARDs or tumour necrosis factor inhibitors, demonstrated that most predictors of remission were representative of rheumatoid arthritis disease severity at baseline, and included male sex, young age, LDA, prior DMARD failure and the absence of rheumatoid factor and anti-CCP.22 Furthermore, a systematic review and meta-analysis comprised mostly of observational studies in patients with rheumatoid arthritis demonstrated that lower baseline disease activity, male sex and a higher education level were predictors of remission, while initial use of corticosteroids reduced the likelihood of remission.23
ORAL Shift open-label phase findings were generally consistent with those observed in the double-blind phase, demonstrating that the efficacy of tofacitinib modified-release 11 mg once daily plus methotrexate was sustained up to week 48 in patients who continued combination therapy.16 Furthermore, the efficacy and safety profiles of tofacitinib modified-release 11 mg once daily in combination with methotrexate appeared similar to the historic profiles of tofacitinib immediate-release 5 mg twice daily in combination with methotrexate.6–8 These findings also demonstrate that the efficacy of tofacitinib modified-release 11 mg once daily plus methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate was generally consistent with that of bDMARDs.24–27
Limitations of this study include the fact that in this open-label phase, patients and investigators were aware of the active treatment, tofacitinib modified-release 11 mg once daily plus methotrexate. Therefore, rates of response, LDA and remission were numerically higher versus a previous randomised, double-blind study in a similar patient population.8 Additionally, because patients were required to achieve LDA (CDAI ≤10) to be eligible for inclusion in the double-blind phase, there may have been an artificial improvement caused by an incentive to stay in the study by the patient and investigator.16 Interestingly, post hoc analyses rates of LDA were numerically lower in the US (68.7%), where access to advanced treatments outside of clinical trials is perhaps greater, versus Europe, Latin America and the Rest of the World (all >90%). However, it should be noted that, overall, response rates were sustained up to week 48 in patients who entered the double-blind phase and continued treatment with tofacitinib 11 mg once daily plus methotrexate,16 indicating that the response rates observed at week 24 of the open-label phase were not primarily artificial. The post hoc analyses of baseline predictors of LDA and remission were exploratory in nature and therefore not designed or powered to detect baseline characteristics predictive of achieving LDA or remission. The multivariable regression model was based on a stepwise procedure, which is at least partially affected by the correlations among the covariates. Finally, it should be noted that this post hoc analysis was conducted at one time point (week 24); therefore, further studies would be required to assess which baseline factors may predict sustained LDA and remission with tofacitinib modified-release 11 mg once daily plus methotrexate.
In all, considering the efficacy and safety profiles demonstrated here for tofacitinib modified-release 11 mg once daily plus methotrexate and previously for tofacitinib immediate-release 5 mg twice daily plus methotrexate in a similar patient population, there are options available in the treatment landscape to accommodate patient preferences. As observed in previous studies and as expected, our data confirm that lower baseline disease activity (CDAI score) generally confers a greater chance of achieving LDA or remission with treatment for rheumatoid arthritis. Previously reported results of the double-blind phase of ORAL Shift indicate that in patients achieving LDA with tofacitinib modified-release 11 mg once daily, withdrawal of background methotrexate may be considered.16 Taking this finding in the context of prior studies demonstrating the effectiveness of tofacitinib as a switch (stopping methotrexate) or add-on treatment in patients with inadequate response to prior methotrexate,8 tofacitinib provides multiple monotherapy treatment regimen options, which allow for patient and physician preference, and facilitate shared decision making between patients and physicians.
Data availability statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Ethics statements
Ethics approval
This study was conducted in accordance with the Good Clinical Practice guidelines of the International Conference on Harmonisation and with the principles of the Declaration of Helsinki. The trial protocol, any amendments, and informed consent documents were reviewed and approved by the Institutional Review Boards and the Independent Ethics Committees at each study centre.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JP, BH, AD, TL and ECK conceived or designed the study. SBC, AD and TL acquired the data. SBC, JP, BH, EM, AD, TL, SL, LS and HS analysed the data. All authors had access to the data, and SBC and SL accessed and verified the data reported in this manuscript. All authors were involved in interpretation of data and reviewed and approved the manuscript’s content before submission.
Funding This study was sponsored by Pfizer Inc. Medical writing support, under the guidance of the authors, was provided by Jennifer Arnold, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, New York, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–4).
Competing interests SBC has served as a consultant and investigator for AbbVie, Eli Lilly, Genentech, Gilead and Pfizer Inc. JP has received research support from AbbVie, Bristol-Myers Squibb, Merck, Roche and UCB; and has acted as a consultant for AbbVie, Actelion, Amgen, Bayer, Bristol-Myers Squibb, Eicos, Eli Lilly, Emerald Pharmaceuticals, Merck, Novartis, Pfizer Inc, Roche, Sandoz, Sanofi, Seattle Genetics, Teva and UCB. BH has received research support or honoraria for advisory board membership or speaking engagements from AbbVie, Amgen, Eli Lilly, Gilead, Merck, Pfizer Inc, Sandoz and UCB. EM has received research support from Eli Lilly, Pfizer Inc and Roche; and has been a member of the speakers’ bureau for AbbVie, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gema Biotech, GlaxoSmithKline, Janssen, Pfizer Inc, Roche, Sandoz and Sanofi. AD, TL, SL, LS, RG, SM and HS are employees and shareholders of Pfizer Inc. ECK has received research support from Amgen, Merck, Pfizer Inc and PuraPharm; has consultancy agreements or advisory board membership with AbbVie, Amgen, Bristol-Myers Squibb, Celltrion, Eli Lilly, F. Hoffmann-La Roche, Gilead, Janssen, Merck, Myriad Genetics, Pfizer Inc, Samsung Bioepis, Sandoz and Sanofi-Genzyme; and has speaker honoraria agreements with AbbVie, Amgen, F. Hoffmann-La Roche, Janssen, Merck, Novartis, Pfizer Inc and Sanofi-Genzyme.
Provenance and peer review Not commissioned; externally peer reviewed.