Article Text

Abstract
Knowledge of pathophysiology of rheumatoid arthritis (RA) has improved over the past decades, which resulted in new treatment options and strategies that led to better clinical outcomes. At the same time, we have come to understand that RA is a heterogeneous disease on a clinical as well as a pathophysiological level. Despite this heterogeneity, current management recommendations still adopt a ‘one-size-fits-all’ treatment approach, where ideally individualised treatment, or personalised medicine, is preferred. The first step towards personalised medicine in RA would be to designate different treatment strategies to distinct clinical or molecular phenotypes of patients. This viewpoint discusses current evidence and elaborates on future possibilities for personalised medicine in RA.
- arthritis
- rheumatoid
- therapeutics
- autoantibodies
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Setting the scene
A young female patient with a newly diagnosed, autoantibody positive rheumatoid arthritis (RA) with functional limitations in daily life visits your outpatient clinic. You decide to start a combination of methotrexate and glucocorticoids. After 3 months, she has an inadequate response to the initial treatment and, therefore, a tumour necrosis factor inhibitor (TNFi) is added. Unfortunately, the disease is still active in the 3 months thereafter, and thus the TNFi is switched to an anti-interleukin 6. Finally, 9 months after diagnosis the patient is in remission. However, what if gene expression, synovial biopsy and/or serum biomarkers, in combination with clinical characteristics, can inform you about treatment choice or response, would you treat this patient differently? What if we knew exactly how to treat, not only her, but every patient with RA?
Introduction
Knowledge of pathophysiology of RA has improved over the past decades, which resulted in new innovative treatment options and strategies that led to better clinical outcomes.1–3 At the same time, we have come to understand that RA is a heterogeneous disease on a clinical as well as a pathophysiological level.1 Despite this heterogeneity, current management recommendations still adopt a ‘one-size-fits-all’ treatment approach, where ideally individualised treatment, or personalised medicine, is preferred.
Personalised medicine is based on identifying subgroups of patients with distinct mechanisms of disease, different prognosis or different responses to treatment. It allows us to develop treatments that are particularly effective for a subgroup of patients. Problems as undertreatment as well as overtreatment and accompanying (serious) adverse events might be circumvented by this approach. In cancer treatment, personalised approaches are most widely used. A well-known example concerns treatment in malignant breast tumours with overexpression of HER2 with the monoclonal antibody trastuzumab.4 5 For RA, personalised treatment based on distinct phenotypes has been proposed in the literature.6 7 Therefore, in this viewpoint we will discuss current evidence and future possibilities for personalised medicine in RA.
Modern management of RA
The following three principles are the foundation of the modern management of RA: (1) early recognition of the disease, (2) early initiation of intensive therapy and (3) a treat-to-target approach. Within this approach, rheumatologists should strive for clinical remission, or low disease activity when remission is not achievable.2 Early recognition and initiation of intensive therapy prevents disease progression and joint destruction.8 If remission is not achieved, treatment will be intensified until the treatment goal is reached, also known as treat-to-target, which has shown to improve short-term and long-term outcomes.9 10
Abovementioned gained knowledge has led to current EULAR and American College of Rheumatology recommendations that advise to start with methotrexate in combination with glucocorticoids as first-line treatment.11 12 If there is an inadequate response to the initial treatment, the absence or presence of poor prognostic factors determine second-line treatment. If poor prognostic factors are present, a biologic or targeted synthetic disease-modifying anti-rheumatic drug (bDMARD or tsDMARD) should be started, usually a TNFi, while another (or a combination of) conventional synthetic (cs)DMARDs should be started if they are absent. These poor prognostic factors include the presence of autoantibodies (anticitrullinated protein antibodies (ACPA) and/or rheumatoid factor (RF)), high disease activity, erosive disease and an inadequate response to ≥2 csDMARDs.11 The choice of second-line treatment based on poor prognostic factors could be regarded as a first step to personalised treatment although evidence supporting this treatment strategy compared with current routine care is limited.
Risk of undertreatment and overtreatment
Still, current treatment initiation and intensifications are largely based on a trial and error approach. As a result, 50%–60% of patients will not reach remission after DMARD initiation and >60% will need ≥3 treatment intensifications to reach the treatment goal.3 Contrastingly, a substantial number of patients with RA do respond to initial or second-line treatments, with sustained remission as a result.3 So, we could argue that very good responders might also do well on less intensive therapies and that inadequate responders, with several treatment intensifications, are (unnecessarily) treated with ineffective medicines, which could be accompanied with (serious) adverse events.
To overcome this risk of overtreatment and undertreatment, and possibly accompanying (serious) adverse events, we need to predict how patients with RA will respond to their therapy. This prediction should preferably take place before start of treatment and otherwise as soon as possible after initiation or intensification, since this increases the chance at achieving remission. Previous studies also showed that reaching remission within 6 months, after the diagnosis of early (rheumatoid) arthritis is made, is associated with better long-term outcomes and, therefore, early treatment prediction will increase the risk at achieving this.13 Dividing early (rheumatoid) arthritis into clinical phenotypes at diagnosis could be the first step to personalised treatment, while molecular biomarkers (and clinical predictors) might assist in treatment choice after an inadequate response (figure 1).
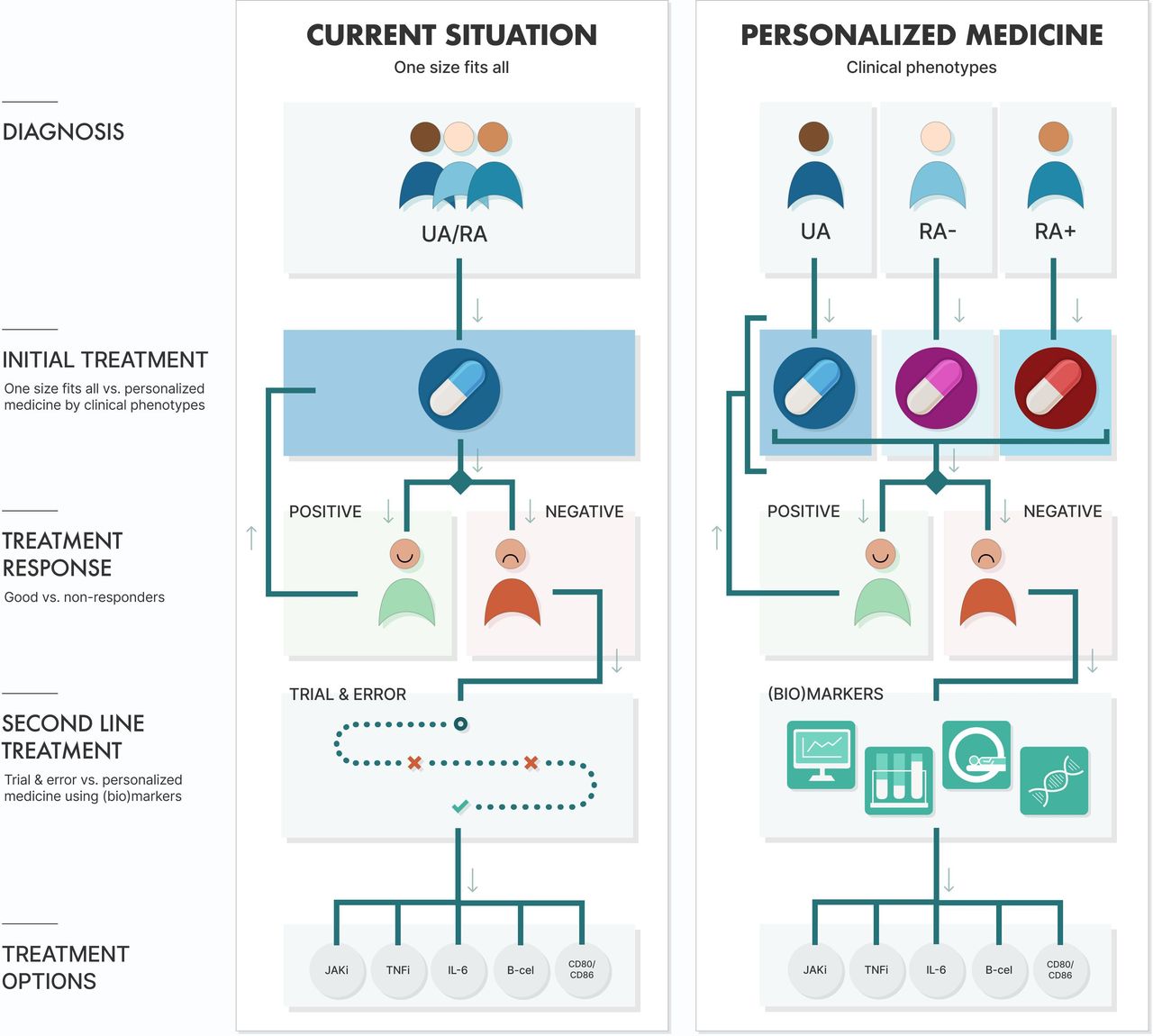
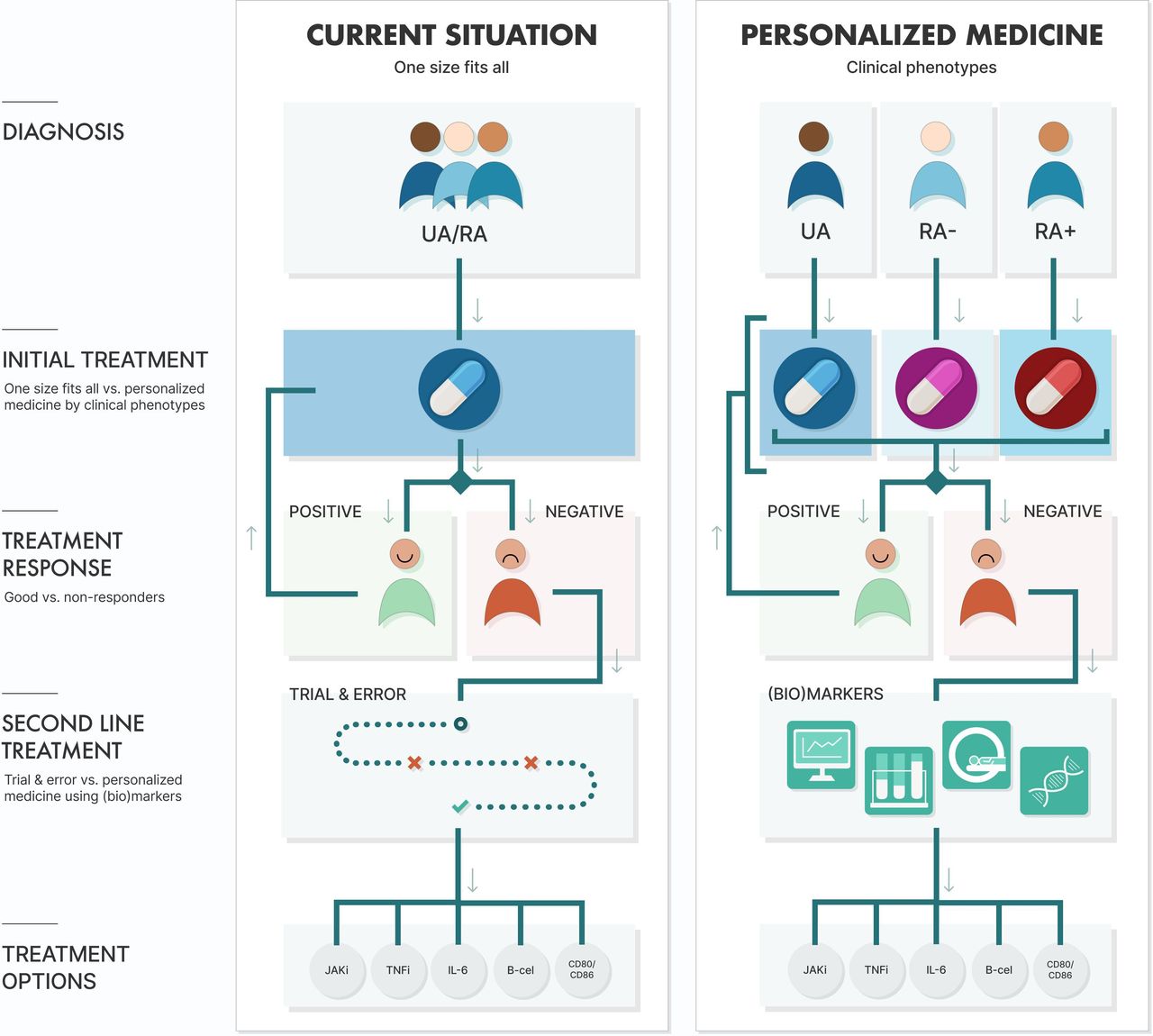{kind=link}
Current management of (rheumatoid) arthritis versus future possibilities for personalised medicine in (rheumatoid) arthritis. Abbreviations: IL-6, interleukin 6 inhibitor; JAKi, Janus Kinase inhibitor; RA, rheumatoid arthritis; RA−, autoantibody negative RA; RA+, autoantibody positive RA; TNFi, tumour necrosis factor inhibitor; UA, undifferentiated arthritis.
Clinical phenotypes of early (rheumatoid) arthritis
Early (rheumatoid) arthritis can be divided into the following three clinical phenotypes: undifferentiated arthritis (UA), autoantibody (ACPA and RF) negative RA (RA−) and autoantibody positive RA (RA+).6 14 UA refers to patients with arthritis who do not meet the 2010 and 1987 classification criteria for RA, which corresponds with the patients with early arthritis described in the EULAR recommendations for early arthritis.15 16 Patients with RA do fulfil either the 2010 or 1987 classification criteria for RA.16 The subdivision of these phenotypes was originally based on differences in (progression of) erosive disease, while nowadays only DMARD-free remission (DFR) rates and the number of treatment intensifications needed to reach remission differ.6 13 14 On the other hand, all clinical phenotypes have a similar impact on patients’ lives when patient-reported outcomes are compared.14 From a pathophysiological point of view, genetic as well as environmental risk factors differ between RA+ and RA−.17–19 Differences in cytokine levels in synovial tissue and fluid between RA+ and RA− have been proposed as well.20 21 Because of the differences in prognosis and pathophysiology treatment might be personalised for these phenotypes, but strategy trials within these subgroups are necessary to validate this.
If we take a closer look at UA then the number of risk factors present determines prognosis. These risk factors are specified in the EULAR recommendations for early arthritis and include (1) the number of swollen joints; (2) elevated acute-phase reactants; (3) presence of autoantibodies and (4) erosive disease.15 Luurssen-Masurel et al, for example, showed that patients with early arthritis with fewer risk factors have a higher change at (sustained) DFR, irrespective of the initial treatment.22 In addition, the authors showed that non-steroidal anti-inflammatory drugs and glucocorticoids are not indicated for this subgroup of patients, but hydroxychloroquine and methotrexate were equally effective.22 However, validation is needed. Fortunately, recently a strategy trial in UA has started that compares different initial treatment strategies Induction of Cure in Early Arthritis trial (I CEA).23 Results of the I CEA trial are expected in 2025.
Also in RA− strategy trials comparing different initial treatment strategies are sparse. Previously, Choi et al showed that treatment response in RA− was better compared with patients with RA+ when given similar DMARD therapy.24 Another trial showed that combination DMARD therapy was only necessary to prevent radiological progression in RA+.25 Finally, a meta-analysis showed that patients with RA+ respond better to rituximab than patients with RA−.26 However, none of these studies compared different treatment strategies in RA−, but recently such a trial was published.7 This study showed a similar efficacy, including disease activity, functional ability and radiological progression, between initial hydroxychloroquine and methotrexate, while glucocorticoids were not indicated for this phenotype.7 However, hydroxychloroquine was better tolerated than methotrexate.7
To summarise, early (rheumatoid) arthritis can be subdivided into three clinical phenotypes, namely UA, RA− and RA+, because of a distinct pathophysiology and different prognosis. These features are prerequisites for personalised treatment, but more importantly growing evidence is showing that UA and RA− can be treated with less intensive treatment compared with RA+.
Treatment choice
In addition to personalised treatment based on clinical phenotype, personalisation can also be done on differences in treatment response. The right choice of DMARD therapy has an important role in obtaining recommended treatment goals, since the time span for the optimal effect is at least 6–12 weeks.27 Therefore, prediction of treatment response is preferably done before start of treatment and otherwise as soon as possible thereafter.
In the near future, biomarkers will probably help rheumatologists in their treatment choice, especially when they have to choose between bDMARDs or tsDMARDs. Tao et al, for example, used machine learning to develop models based on gene expression and DNA methylation data to predict response to adalimumab and etanercept in patients with RA.28 They reported a response prediction with an accuracy of 85.9% (adalimumab) and 79% (etanercept) when using gene expression and 84.7% (adalimumab) and 88% (etanercept) when using DNA methylation. Another study used a combination of clinical features and RNA sequencing to predict response to TNFis.29 Finally, Humby et al published a trial that applied a stratified approach based on histology and RNA-sequencing of synovial biopsies to compare the effectiveness of rituximab and tocilizumab in patients with RA with an inadequate TNFi response.30 The authors showed that in patients with RA with a low or absent B-cell lineage expression, tocilizumab was more effective than rituximab. Another biopsy-driven trial Stratification of Biologic Therapies for RA by Pathobiology trial, in patients with RA with an inadequate response to csDMARDs, is being conducted.31 Studies like these are necessary since patients tend to have the longest drug survival on their first bDMARD.32 In addition to these trials, the ongoing observational PRECISE-RA (PRECISion medicinE Across the Disease Continuum to Prevent and Treat Rheumatoid Arthritis) study collects blood, urine and synovial samples in order to prevent and treat RA with personalised medicine approaches.33
An alternative for prediction of treatment response at start would be prediction as soon as possible thereafter. Advantages of this approach are that one can look at changes in biomarkers. Miyazaki et al showed that TNF-α levels 24 hours after initiation of certolizumab were associated with a higher chance at treatment response in the 3 months thereafter.34 Clinical factors can also be used as predictors. For example, the early glucocorticoid response, measured within 1 month, is a useful tool for recognising patients with RA who will probably fail on their initial csDMARD strategy.35 In addition, Kume et al showed that improvement in sonography measures 2 weeks after tocilizumab initiation could predict treatment response after 24 weeks.36
To summarise, prediction of treatment response at initiation or as soon as possible thereafter is vital for improving remission rates and/or drug survival . Fortunately, more and more evidence is emerging which might help the treating rheumatologist to choose the most effective treatment in the near future.
Future possibilities
Although the importance of personalised medicine is recognised, the implementation in daily practice is hampered, due to lack of evidence. Therefore, clinical trials aimed at personalised medicine as well as translational research targeted at finding molecular biomarkers are needed to identify subgroups that will or will not respond to a certain treatment strategy.37 Some possibilities are, for example, randomised clinical trials (RCTs) that compare different treatment strategies within aforementioned clinical RA phenotypes or RCTs that compare standard of care with personalised treatment strategies in which early treatment adaptions are based on (changes in) biomarkers, for example, Certolizumab switch after 1 gift if TNF-α levels are too high, which is derived from the study of Miyazaki et al.34 From a translational point of view, personalised treatment in RA requires a better understanding of molecular mechanisms and the relationship with response to different therapies. Together, these would be the next steps towards personalised medicine, wherein problems as undertreatment as well as overtreatment and accompanying (serious) adverse events are circumvented.
Conclusion
In conclusion, healthcare is more and more shifting towards a patient-centred care approach. Therefore, in our opinion, the management of early (rheumatoid) arthritis should undergo the same changes. With the wide range of available treatment options and the need for early effective treatment, personalised medicine should be our priority. Existing evidence and ongoing studies provide us with hope that the implementation of personalised medicine in daily practice for RA will become available in upcoming years. We are, therefore, confident that new data, together with existing evidence, will soon lead to personalised treatment of our newly diagnosed patient with RA.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank J.T. Venbrux for his help in preparing the figure.
References
Footnotes
Contributors Both authors contributed to conceptualisation and writing. JH contributed to figure and PHPdJ contributed to supervision.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.