Article Text

Abstract
Objective To explore at the molecular level the phenotype of a patient suffering an autoinflammatory syndrome which was diagnosed as familial cold autoinflammatory syndrome type 2 (FCAS-2). To explore the functions of Nlrp12 in inflammation using mouse models.
Methods Whole exome sequencing and Nlrp12 targeted resequencing were performed on DNA isolated from the patient and her family members. In vivo and ex vivo models of inflammation (urate crystals-dependent acute joint inflammation and urate crystals-induced peritonitis) were analysed in Nlrp12-deficient and Nlrp12-competent mice.
Results A rare missense NLRP12 variant (c.857C>T, p.P286L) was identified in the patient and her healthy relatives. Nlrp12-deficient mice exhibit reduced systemic inflammation and neutrophilic infiltration.
Conclusion Nlrp12 mediates proinflammatory functions in mice. In humans, the identification of Nlrp12 variants must be cautiously interpreted depending on clinical and paraclinical data to diagnose FCAS-2.
- inflammation
- autoimmune diseases
- rheumatic fever
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
In humans, several rare mutations in NLRP12 have been associated to autoinflammatory syndromes.
What does this study add?
This study reports the case of a rare NLRP12 variant in both healthy and affected family members, suggesting low penetrance/variable manifestation of the mutation.
How might this impact on clinical practice or further developments?
Diagnosis of familial cold autoinflammatory syndrome type 2, an autoinflammatory disease exhibiting considerable phenotypic variation, requires both genetic and clinical (therapeutic response to Il-1β blockers) information.
Introduction
Systemic autoinflammatory disorders (SAIDs) are caused by defects of innate immune responses leading to excessive inflammation via many different pathways.1 Patients share recurrent flares of fever, dramatic elevation of acute phase reactants and variable clinical manifestations such as rash. Recently, a new classification (including genetic data) of the these defects, also called hereditary recurrent fever syndromes, has been published.2 Among these diseases, inflammasomopathies are the most common.3 Inflammasomes are intracellular multiprotein complexes involved in sensing and responding to both extracellular and intracellular factors, leading to the maturation and release of bioactive interleukin (IL)-1β which is necessary for appropriate responses against various pathogens.4 These complexes are formed on ligand-dependent association of a sensor, the adaptor ASC (Apoptosis-associated speck like protein containing a caspase recruitment domain) (except in the case of the NLRP1 and NLRC4) and the procaspase 1 which becomes activated by proximity-induced self-cleavage.5 Many sensors have been described, belonging to either the NLRs (nucleotide-binding domain and leucine-rich repeat containing receptors), absent in melanoma 2-like receptors and pyrin families, but the most extensively studied is NALP3, which forms the NLRP3 inflammasome. Together with Pyrin (306 unique variants reported in the Leiden Open Variation Database), NALP3 (235 variants) is a frequently affected inflammasome, causing respectively cryopyrinopathies (or cryopyrin-associated period syndromes) and familial Mediterranean fever.1 6 In contrast, NLRP12 plays controversial functions, as it can assemble into an inflammasome or act as a negative regulator of inflammation.7 Its involvement in host defence appears undisputable, at least in mice,8 but whether variants in the NLRP12 gene in humans are associated with autoinflammatory syndromes is still debated.1 To date, 57 unique variants have been described in patients who exhibit considerable clinical heterogeneity.9 10 Nevertheless, familial, cold, autoinflammatory syndrome type 2 (FCAS-2, OMIM # 611762) is depicted as a rare condition associated with NLRP12. Since the initial description of the first two patients,11 61 cases with NLRP12 variants have been reported so far.9 Altogether, it appears that NLRP12, which ligand remains unknown, carries multiple immune functions, still elusive in the present state of our knowledge.12
Here, we describe a rare missense variant in NLRP12 in a patient with late onset autoinflammatory syndrome that was successfully treated with IL-1 blockade. These clinical observations suggesting a potential role of NLRP12 in autoinflammation were further supported by in vivo and ex vivo investigations of mouse models. However, further genetic analyses revealed that this variant was also present in healthy family members of the proposita, indicating a possible incomplete penetrance and supporting a controversial role of NLRP12 in autoinflammation.
Methods
Genetic analyses
For whole exome sequencing (WES), genomic DNA was isolated from patient and parents’ peripheral blood or saliva using standard protocols. Exome sequencing was performed on a NextSeq500 sequencer (Illumina, San Diego, California, USA) as described previously.13 We focused only on protein-altering variants (missense, nonsense, splice site variants and indels). To identify potential causal variants, we further filtered the variants based on a dominant mode of inheritance, that is, variants present in the patient and absent in her healthy daughter. For targeted resequencing, the candidate variants were confirmed by capillary Sanger sequencing on an ABI PRISM 3730xl sequencer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and analysed on the SeqScape software (Thermo Fisher Scientific).
Quantification of cytokines
IL-1β(ref no 88-7261-22), IL-6 (ref no 88-7066-22) and IL-8 (ref no 88-8086-88) were quantified using dedicated ELISA according to the Manufacturer’s recommendations (eBioscience, San Diego, California, USA).
Real-time quantitative PCR
Total RNA was prepared from joints (dissected hindpaws) as described in14 and reverse transcribed using the cDNA synthesis kit (iScript ready-to-use cDNA supermix, Biorad, ref no 4106228). Real-time quantitative PCR (RT-qPCR) was performed in a total volume of 20 µL using the Sso-advanced universal SYBR-Green supermix (Biorad, ref no 10000076382) and gene-specific primers (list available on request). After a denaturing step at 95°C for 30 s, 40 cycles were performed (95°C for 5 s and 60°C for 20 s) using a Rotor-Gene 6000 RT-PCR machine (Corbett Life Science). Results were obtained using the SDS Software (Perkin Elmer). Melting-curve analysis was performed to assess the specificity of PCR products. Relative expression was calculated using the comparative threshold cycle (Ct) method. The Ct of the gene of interest was adjusted to the average Ct of three housekeeping genes (Hprt, Actin and Gapdh) to obtain a ΔCt.
Statistics
Following normality tests (Kolmogorov-Smirnov and Shapiro-Wilk), data were analysed with a Mann-Whitney test or unpaired t test (two-tailed unpaired) to compare two independent groups and non-parametric (Spearman) test for correlation analysis. Statistics were calculated with GraphPad V.5.01 software. A p<0.05 was considered significant. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Results
Case presentation
The patient is a 75-year-old French woman (born 1944 of Italian parents) who was diagnosed with rheumatoid arthritis in 2008 (with rheumatoid factor, antinuclear antibodies), a benign cutaneous mastocytosis and osteoporosis without fracture. No long-term therapy was initiated. In December 2011 (at the age of 67), she suffered periodic fever (38°C–38.8°C) occurring in the morning twice or three times per week, associated with diffuse myalgia, chronic urticaria (that lasted 4 months and was not cold-dependent), unilateral deafness and a deterioration of the general state. Joint examination revealed no synovitis, enthesitis or dactylitis. Her cardiac and pulmonary examination was within normal limits. Laboratory data revealed increased inflammatory markers (neutrophilia, C reactive protein, CRP above 100 mg/L), antinuclear antibody positive (1/640) with dense speckled fluorescence, native-DNA antibody and rheumatic factor slightly positive, anti extractable nuclear antigen negative; analyses of complement components and Creatine Kinase (CK) were normal. Q fever serology was slightly positive (treatment with doxycycline was not contributing); other analyses (including blood cultures collected during fever peaks) for potential infectious diseases (brucellosis, borreliosis, rickettsiosis, HIV, hepatitis B, hepatitis C, leptospirosis) were negative. Positron-emission tomography, transthoracic echocardiography and bone marrow biopsy were normal. Muscular biopsy showed mild non-specific inflammatory infiltrates. Steroids and methotrexate did ameliorate her symptoms but had no effect on fever and the inflammatory syndrome persisted (CRP at 40 mg/L). An autoinflammatory syndrome was evoked. She finally received IL1-blockers (anakinra first, that was stopped because of induced urticaria, followed by canakinumab, once a month, then once every 2 months) quickly resulting in a full clinical and biological recovery.
Genetic investigations
All the individuals in which genetic investigations were realised in this study gave their written informed consent to participate and for publication.
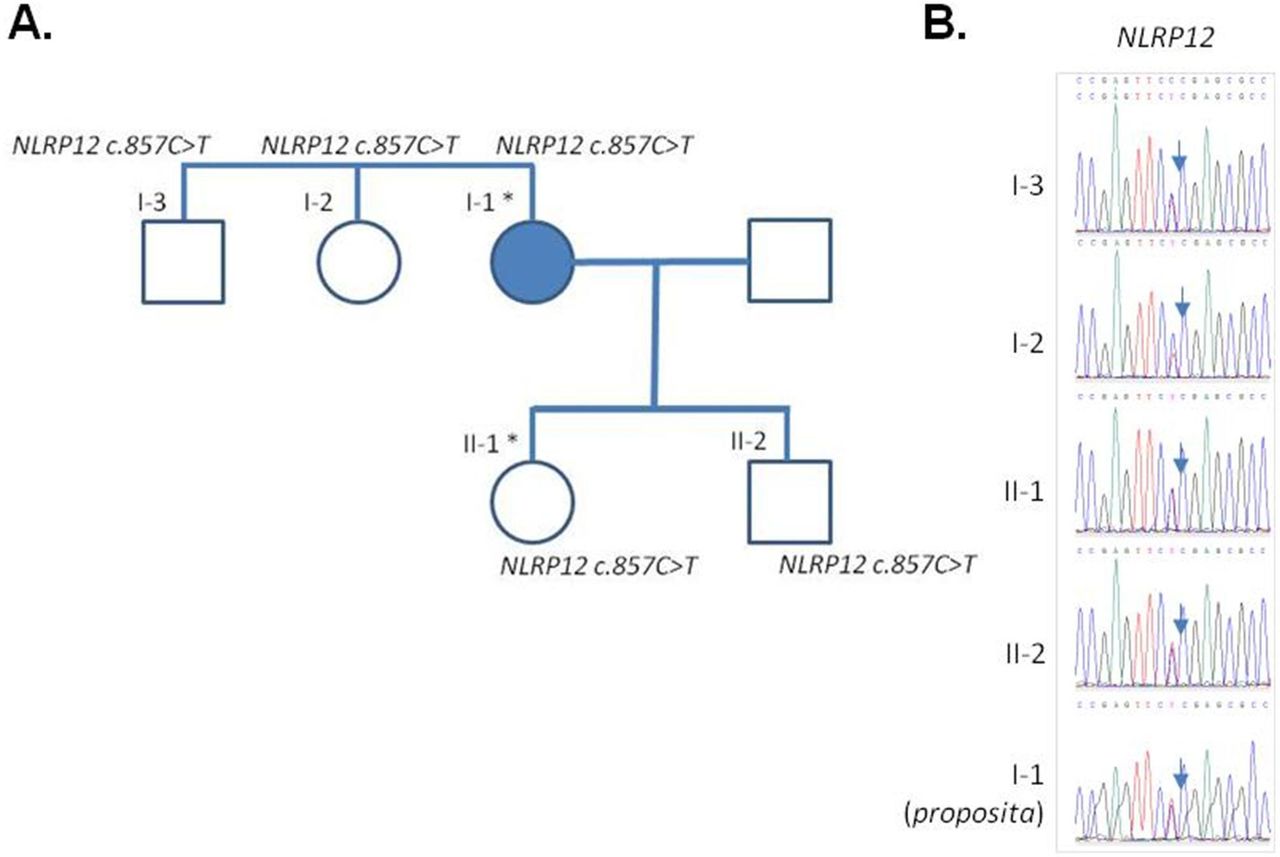
At this stage, an autoinflammatory syndrome was considered and genetic analysis involving the targeted resequencing of all the exons in a panel of 32 genes involved in autoinflammatory diseases (online supplemental table 1) was performed. This revealed the presence of a heterozygote variant (c.857C>T, p.P286L) in a conserved domain of NACHT, LRR and PYD domains-containing protein 12 (NLRP12) (online supplemental figure 1) which has not been described so far; the patient was diagnosed a NLRP12-associated periodic syndrome (NAPS12)15 with late onset. However, because the association of NLRP12 variants and inflammatory syndrome is still questioned,1 a WES was performed on the patient’s DNA and her unaffected daughter (born in 1974, aged 46 when blood was collected). We analysed all variants and found no pathogenic variants in an extended list of 55 genes related to inflammasome activation (a compilation of the genes included in the re-sequencing panel, of the fever panel described in16 and of additional inflammasome-related genes, online supplemental table 1). This analysis confirmed, in the patient’s exome, that the very rare (allele frequency=0.0001554 in the GnomAD database) variant of NLRP12 was the only candidate variant that could possibly explain the phenotype. However, we discovered that the unaffected daughter inherited the c.857C>T variant in NLRP12; further genetic analyses (consisting in a targeted resequencing of NLRP12, and not a WES) of additional family members revealed that this variant is also present in the proposita’s son, brother and sister (figure 1A,B), in which nor clinical items of autoinflammatory syndrome/FCAS2 (fever, rash, urticaria, arthralgia, myalgia, headache), neither biological signs of inflammation were apparent. We interpreted these data as an additional example of possible low penetrance of NLRP12 variants, as recently discussed.17
Supplemental material
Supplemental material

Pedigree and validation of the NLRP12 variant. (A) Family tree. The full blue symbol indicates the affected individual (I-1, the proposita). An asterisk denotes the individuals in which whole exome sequencing was performed. (B) Sanger sequencing electropherograms of the variant for the different family members. IL-2, interleukin 2.
Mouse experiments
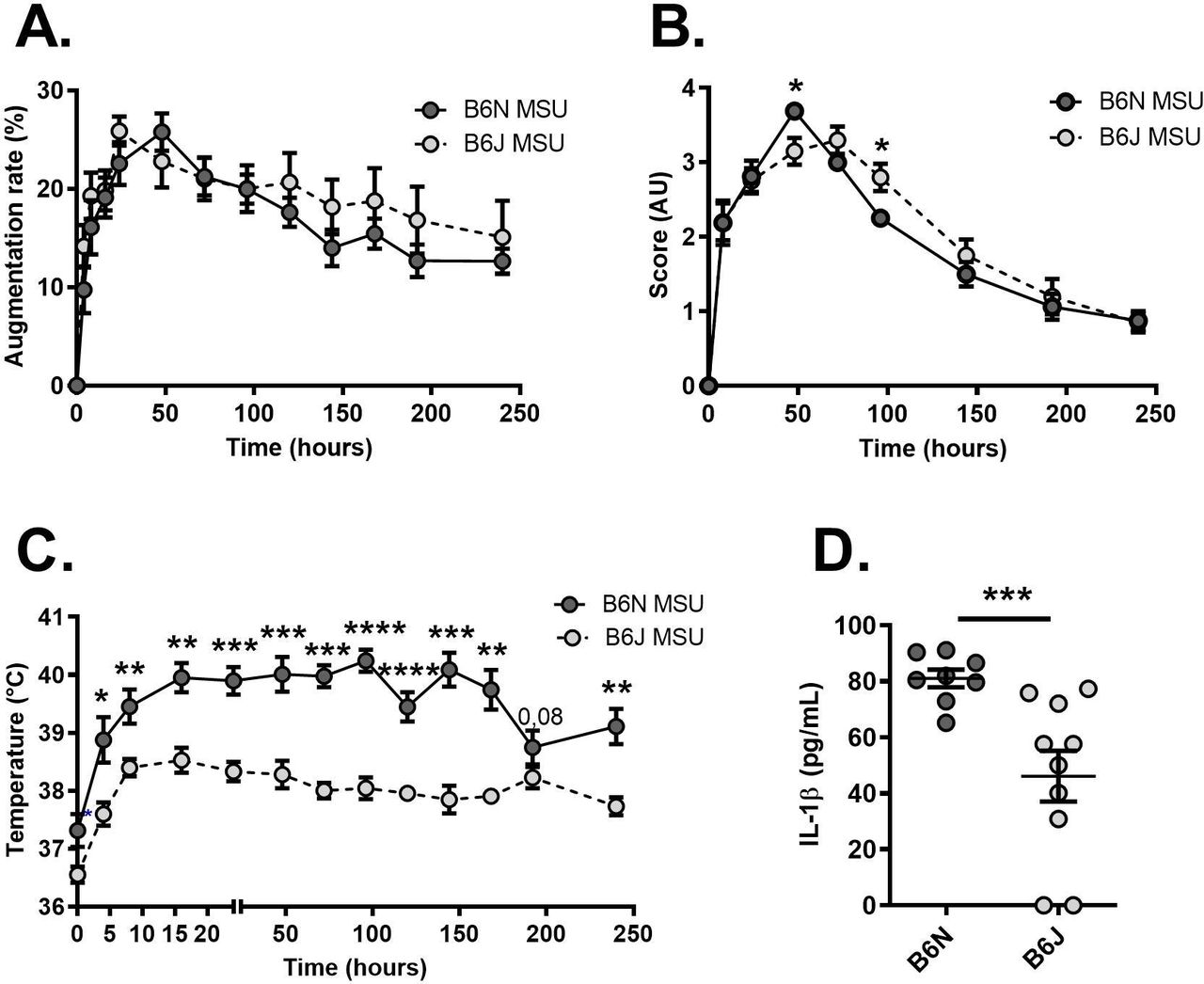
To better describe the potential role of NLRP12, we used mouse models of inflammation. For this, we compared the phenotype of C57Bl/6N (B6N) mice (here used as controls harbouring a wild-type NLRP12 gene) to that of C57Bl/6J (B6J) animals which have a spontaneous missense mutation (inducing a p.R1034K change at the protein level) that is phenocopied by the deletion of NLRP12.18 We first induced an acute inflammatory response in the joint, mimicking gouty arthritis, by subcutaneous injection of monosodium urate (MSU) crystals.14 As seen in figure 2, no difference was seen between controls (B6N) and NLRP12-deficient (B6J) mice with regard to paw thickness (figure 2A) and the clinical score reflecting redness and oedema of the joint (figure 2B). However, we noticed that only a moderate increase in the body temperature could be recorded in C57Bl/6J (B6J) mice on MSU crystals injection, as opposed to controls (figure 2C). This observation, along with reduced seric IL-1β in B6J animal 120 hours after MSU injection (figure 2D), suggests that NLRP12 likely promotes systemic responses to an inflammatory trigger.
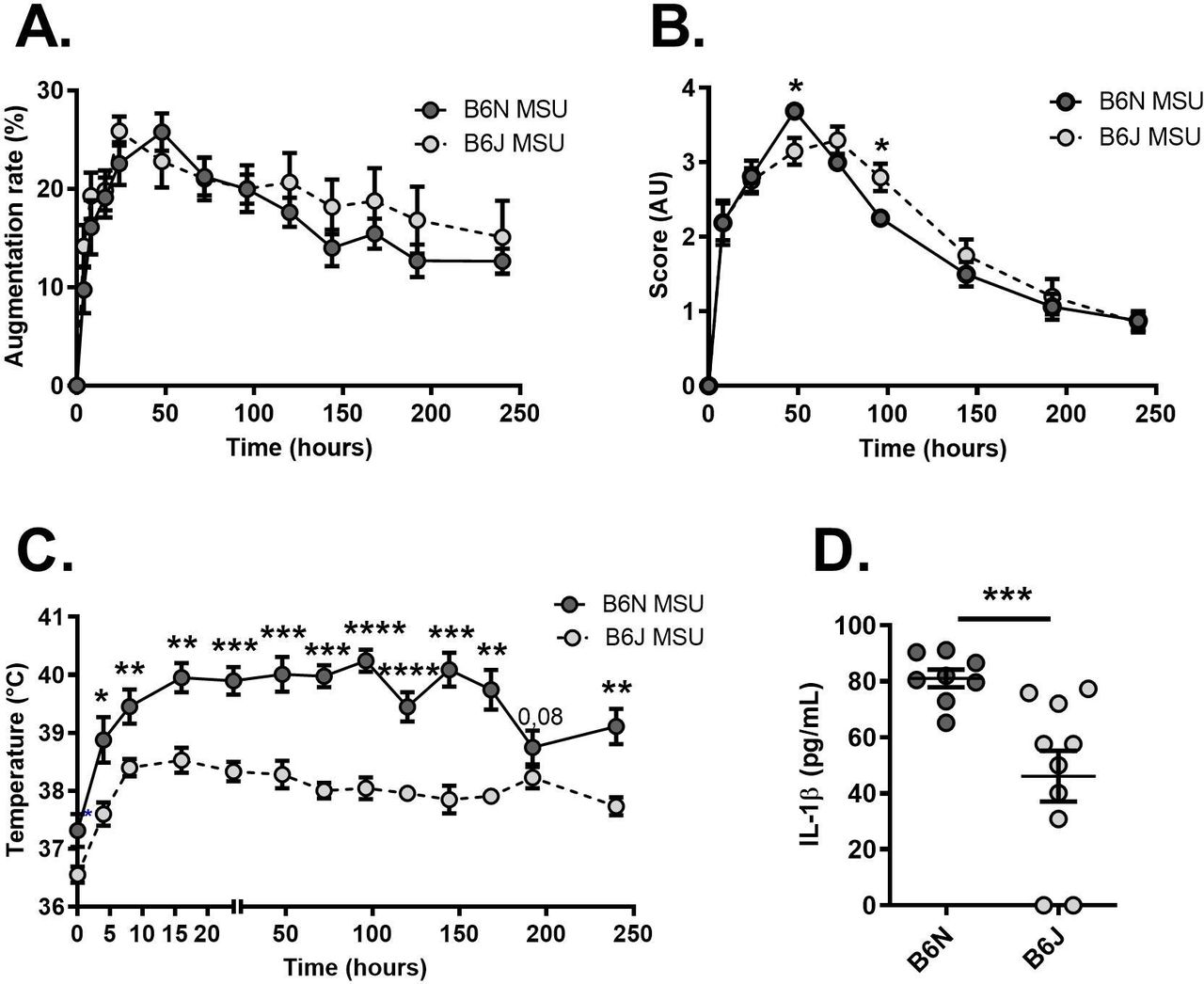
Reduced fever in Nlrp12-deficient mice on MSU crystal-dependent acute joint inflammation. (A) Following monosodium urate (MSU) crystals injection in the paws, the size of the Ankles was measured during 1 day with a calliper in C57BL/6N (B6N controls, N=8 shown in dark grey) and C57BL/6J (B6J Nlrp12-deficient, N=10 show in light grey) mice. The augmentation rate is expressed as a percentage of the ankle size measured at day 0 before MSU injection. (B) The clinical score, reflecting both redness and swelling was evaluated in the same animals and is expressed in arbitrary units (AU). (C) Recording of the body temperature in the same mice. (D) Quantification by ELISA of seric IL-1β 120 hours after MSU crystals injection in controls (B6N) and Nlrp12 mutants (B6J) mice. Data were analysed with a Mann-Whitney U test. *P<0.05. **p<0.01, ***p<0.001. IL-1β, interleukin 1β
We next used a model in which peritonitis is induced by injection of MSU crystals into the peritoneal cavity. In this setting, while the total number of cells in the peritoneal fluid obtained 6 hours after MSU injection remained similar in controls (B6N) and Nlrp12 mutants (B6J, online supplemental figure 2A), we noted reduced neutrophilic infiltration in Nlrp12-deficient mice (online supplemental figure 2B,C). Quantification of inflammatory cytokines IL-6 and TNF remained similar in the peritoneal fluid of both strains (online supplemental figure 2D,E). These data suggest that Nlrp12 participates in the development of an inflammatory setting by promoting neutrophil migration.
Supplemental material
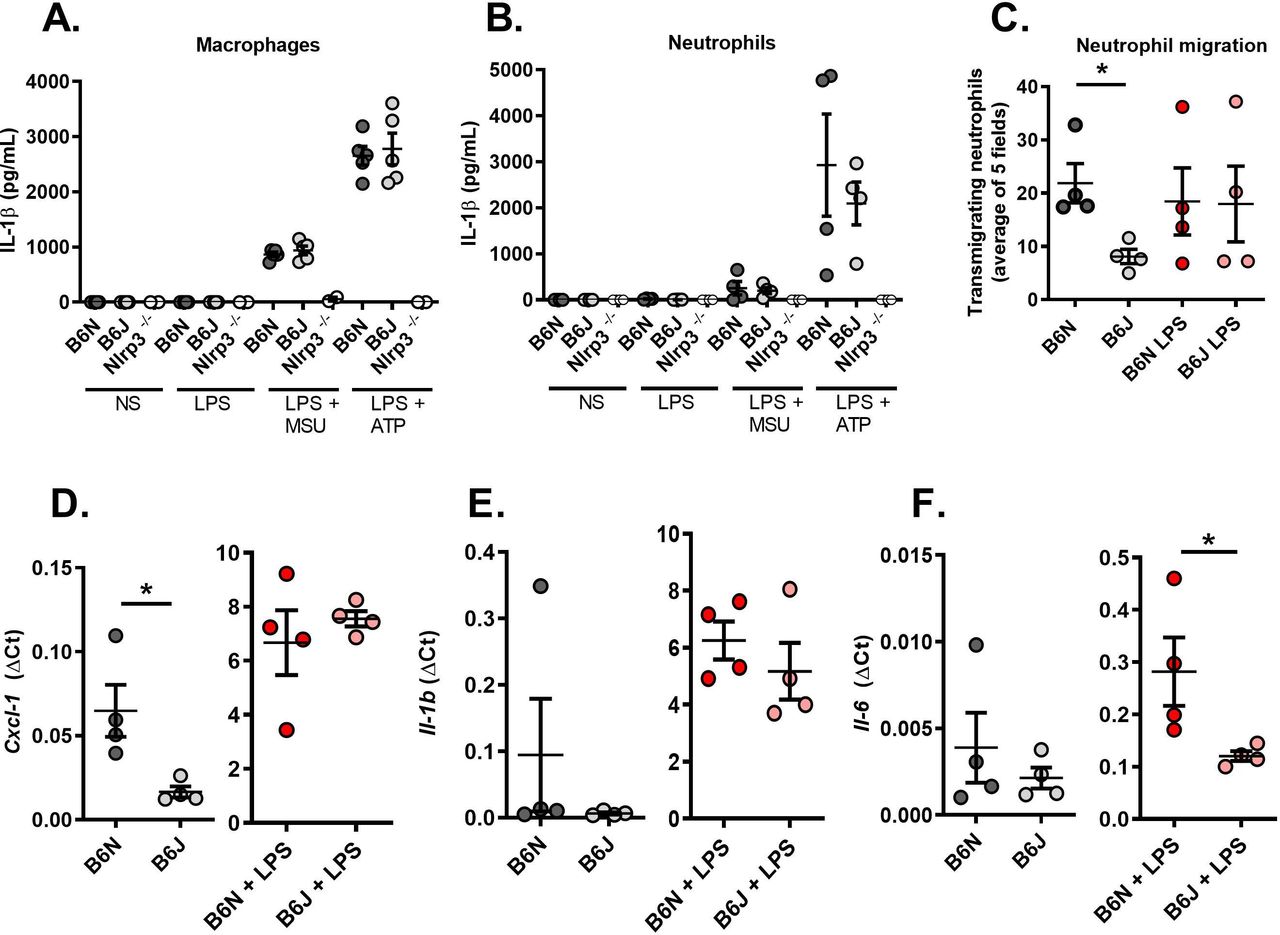
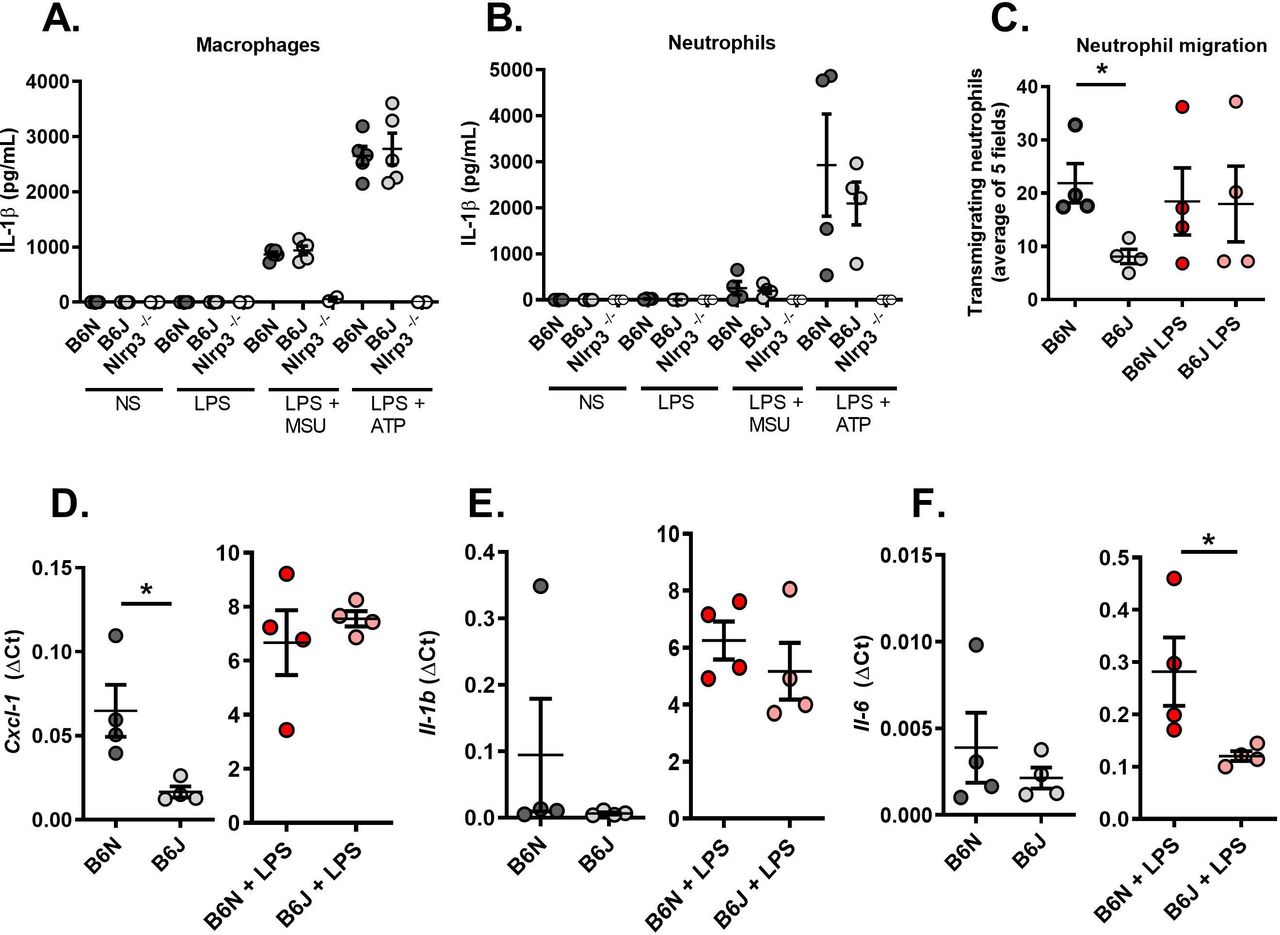
Finally, we turned to an ex vivo model using primary thioglycolate-elicited macrophages and neutrophils harvested from C57Bl/6N and J mice to better understand Nlrp12-dependent molecular mechanisms at the cellular level. We first observed that Nlrp12 deficiency in B6J mice does not impair IL-1β secretion by stimulated macrophages or neutrophils (following Lipopolysaccharide (LPS) and MSU or LPS and Adenosine triphosphate (ATP) addition in the culture medium, figure 3A,B). To analyse the interactions between these cells, we used Boyden chambers and quantified the proportion of neutrophils migrating towards macrophages that are placed inside the well in 24 hours. As seen in figure 3C, cells harvested from B6J mice exhibit reduced migration. Of note, LPS stimulation abolished the difference between controls (B6N) and Nlrp12-deficient cells (B6J). This is in line with reduced Cxcl1 (encoding a major neutrophil chemoattractant) expression in steady-state macrophages isolated from B6J mice; again, LPS stimulation normalised gene expression compared with control B6N cells (figure 3D). Finally, we noted that Il-1β expression by resting or stimulated macrophages remained similar whether Nlpr12 is functional or not (figure 3E) and that LPS-stimulated B6J macrophages expressed reduced Il-6 (figure 3F), which may account for the low fever seen on MSU crystal-induced arthritis in these animals.
{kind=link}
{kind=link}
{kind=link}
Resting Nlrp12-deficient macrophages exhibit reduced neutrophil chemoattractant activity. (A) Macrophages and (B) neutrophils from controls (B6N, N=5) and Nlrp12-deficient (B6J, N=5) mice exhibit similar responses on inflammasome activation (LPS +MSU and LPS +ATP). Nlrp3-deficient cells (N=2) were used as negative controls. (C) Neutrophil (expressed as the mean of 5 microscopic fields) attraction by macrophages was evaluated using a Transwell system. Cells from control (B6N) or mutant (B6J) mice were either left untreated or stimulated with LPS for 24 hours. (D) Cxcl1, (E) Il-1β and (F) Il-6 gene expression was quantified by RT-qPCR in control (B6N) and mutant (B6J) macrophages with or without LPS stimulation. Relative gene expression is indicated as ΔCt (ratio between the CT of the gene of interest and the average of the CTs of 3 housekeeping genes). IL-6, interleukin 6; MSU, interleukin; RT-qPCR, real-time quantitative PCR.
Discussion
Our observations in mouse models indicate that Nlrp12 plays important roles in neutrophil recruitment, as noted previously.18 Given the prominent role of these cells in sterile inflammation, we suspect that their limited recruitment in Nlrp12-deficient C57BL/6 J mice participates in the reduced inflammatory responses in vivo in these animals (figure 3 and online supplemental figure 2).
Supplemental material
Inflammation is a complex process conferring protective immunity against a vast array of pathogens. To fulfil this task, inflammation requires many pathogen recognition receptors which have been selected throughout evolution, and trigger multiple signalling pathways driving innate and adaptive immunity. This response needs to be highly controlled, as evidenced in several instances of excessive inflammation driving tissue damage and chronic diseases. In the past 10 years, huge progresses have been made in the identification of genes involved in autoinflammatory diseases. With the advent of Next-Generation Sequencing technologies, the number of genes associated with autoinflammatory diseases has grown to 60. However, many of these diseases remain difficult to diagnose because of their heterogeneous clinical presentation. Furthermore, genetic analyses are difficult to interpret because a large number of variants in inflammatory genes have been described, some of which are included in genetic panels, while others, of ‘unknown significance’, are not considered. Hence, predictions of autoinflammatory syndromes based on genetic analyses remain challenging. This is particularly true for inflammasomopathies, a rare subset of SAIDs exhibiting a wide spectrum of phenotypes.3 Among them, NLRP12-associated autoinflammatory disease (also known as familial cold autoinflammatory syndrome 2) is a recently described entity essentially based on the presence of arthralgia, periodic fever in patients without canonical mutations in NLRP3.11 To date, few such patients were identified (see ref. 17 for the most recent description) but the association between NLRP12 variants and spontaneous inflammation is not yet firmly established, nor is the molecular role of this inflammasome in the establishment of systemic inflammatory settings.1 9
In this work, we described a patient with late onset auto-inflammatory syndromes which was diagnosed a likely FCAS2 based on the identification, by targeted gene resequencing, of a NLRP12 variant of unknown significance (although several bioinformatics programmes suggest potential damaging consequences of this mutation, see online supplemental table II) which has not been ascribed so far as causal for any disease. Consequently, Anakinra was prescribed and resulted in a complete remission, including fever extinction and CRP negativation. Following cutaneous reaction, Canakinumab was preferred, with similar results and better tolerance. IL-1β overproduction and inflammasome overactivation are hence likely responsible for the symptoms observed in this patient. WES performed secondarily confirmed that the NLRP12 variant was the best candidate variant to explain the phenotype but also unexpectedly showed that it was inherited by her (presently) healthy daughter. Furthermore, targeted NLRP12 resequencing showed that the son, the brother and the sister of the proposita share the same variant, without experiencing any sign of inflammation. These data are more in favour of a low penetrance of this variant.9 17 Furthermore, the pleiotropic and context-dependent functions of NLRP1219 combined with environmental factors (nutrition, microbiota, history of infections and vaccinations) may also explain the clinical heterogeneity among individuals carrying NLRP12 variants.
Our investigations in mouse models, both in vivo and ex vivo, confirmed the prominent role of NLRP12 in neutrophil recruitment (or increased differentiation), which, following NETosis,20 may trigger IL-1β-dependent inflammatory episodes. In agreement with our observation of NLRP12-dependent expression of Cxcl1 in mice, we also noted slightly increased secretion of the neutrophil chemotactic factor IL-8 by primary monocytes isolated from our patient (cells were isolated before Canakinumb injection and 2 months after the previous one), compared with cells harvested from her daughter or healthy donors (online supplemental figure 3A). In contrast, we detected similar levels of IL-1β secreted by the patient’s and her daughter’s monocytes (online supplemental figure 3B). These data should, however, be cautiously interpreted, as our gene expression quantification in mouse cells performed by RT-qPCR, may be not comparable to cytokine quantification performed by ELISA in the supernatant of culture human cells.
Supplemental material
Altogether, our work in mouse models highlights the importance of Nlrp12 in neutrophil-dependent inflammation, while evidence for a role of NLRP12 in human inflammasomopathies remains equivocal.
Ethics statements
Patient consent for publication
Ethics approval
The study as approved by institutional ethics committees. All participants signed an informed consent to participate in the study and for publication. The work on human samples has been approved by the Comité de Protection des Personnes Est IV (approval no 13/48). Animal experiments were reviewed by institutional ethic committee (CREMEAS, approval number 2018083014133041).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DL, AMa, AD, CM, AP and AMo performed experiments. DL, AMa, AD, AMo, RC, AMe and PG analysed data. FM performed the initial diagnosis and recruited the patient. DL, RC and PG wrote the manuscript. All the authors participated in proofreading the manuscript and approved the final version.
Funding Work in our laboratory is supported by the Strasbourg’s Interdisciplinary Thematic Institute (ITI) for Precision Medicine, TRANSPLANTEX NG, as part of the ITI 2021-2028 programme of the University of Strasbourg, CNRS and INSERM, funded by IdEx Unistra (ANR-10-IDEX-0002) and SFRI-STRAT’US (ANR-20-SFRI-0012), the INSERM UMR_S 1109, the University of Strasbourg (IDEX UNISTRA), the European regional development fund (European Union) INTERREG V program (project PERSONALIS) and the MSD-Avenir grant AUTOGEN. The experiments on human cells were partly funded by Sobi France.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.