Article Text

Abstract
Objectives Fatigue is common and severe in primary Sjögren’s syndrome (pSS). The aim of this study was to identify genetic determinants of fatigue in pSS through a genome-wide association study.
Methods Patients with pSS from Norway, Sweden, UK and USA with fatigue and genotype data available were included. After genotype imputation and quality control, 682 patients and 4 966 157 genetic markers were available. Association analysis in each cohort using linear regression with fatigue as a continuous variable and meta-analyses between the cohorts were performed.
Results Meta-analysis of the Norwegian and Swedish cohorts identified five polymorphisms within the same linkage disequilibrium block at the receptor transporter protein 4 (RTP4)/MASP1 locus associated with fatigue with genome-wide significance (GWS) (p<5×10−8). Patients homozygous for the major allele scored 25 mm higher on the fatigue Visual Analogue Scale than patients homozygous for the minor allele. There were no variants associated with fatigue with GWS in meta-analyses of the US/UK cohorts, or all four cohorts. RTP4 expression in pSS B cells was upregulated and positively correlated with the type I interferon score. Expression quantitative trait loci effects in whole blood for fatigue-associated variants at RTP4/MASP1 and levels of RTP4 and MASP1 expression were identified.
Conclusion Genetic variations at RTP4/MASP1 are associated with fatigue in Scandinavian pSS patients. RTP4 encodes a Golgi chaperone that influences opioid pain receptor function and MASP1 is involved in complement activation. These results add evidence for genetic influence over fatigue in pSS.
- polymorphism
- genetic
- sjogren's syndrome
- autoimmune diseases
Data availability statement
Data are available on reasonable request. Genotype data are available on request from the authors on a collaborative basis.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Fatigue is a common phenomenon in chronic inflammatory diseases including primary Sjögren’s syndrome (pSS).
Genetic and epigenetic studies indicate a genetic component of fatigue.
What does this study add?
Our study demonstrates that genetic variants are associated with fatigue in pSS and specifically that the major allele of the indel rs60344347 at the receptor transporter protein 4 (RTP4)/MASP1 locus was associated with higher fatigue in Scandinavian patients with pSS.
RTP4 encodes a protein involved in pain perception and may represent a functional link between pain and fatigue.
How might this impact on clinical practice or further developments?
Insight into genetic susceptibility to fatigue encourages a better understanding of the phenomenon, which may facilitate improved patient education, possibilities for precision medicine and eventually development of targeted treatment.
Introduction
Primary Sjögren’s syndrome (pSS) is a chronic systemic autoimmune disease. Fatigue, described as ‘an overwhelming sense of tiredness, lack of energy and feeling of exhaustion’, affects 70%–80% of pSS patients and causes a marked reduction of quality of life.1–3 The exact mechanisms leading to fatigue are unknown. Activation of innate immunity and proinflammatory cytokines are recognised as important factors.4 5 Evolutionarily, fatigue can be considered to be a part of sickness behaviour, a protective behaviour that increases the chances of survival during acute sickness.6 This behaviour turns inexpedient in states of chronic inflammation.
Fatigue seems to persist in the individual pSS patient.3 7 This persistence, as well as the observation that patients with similar disease activity often report large differences in fatigue severity, could indicate that genetic variation influences fatigue. Polymorphisms in genes encoding tumour necrosis factor-α, interleukin (IL)1β, IL4 and IL6 have been associated with fatigue in various conditions.8 9 We have previously reported that genes involved in regulation of innate and adaptive immunity are differentially methylated in pSS subjects with high vs low fatigue.10 Our hypothesis is that specific genes contribute to regulation of fatigue and that genetic variation can influence fatigue severity. Consequently, we sought to identify genetic variants implicated in fatigue in pSS through a Genome-Wide Association Study (GWAS).
Materials and Methods
Patient and public involvement statement
Patients were not involved in the planning or conduction of the study.
Subjects
The patients were part of a larger GWAS of pSS,11 and the 690 patients with both genotype and fatigue data available were included in this study (online supplemental figure S1). All patients fulfilled the American-European Consensus Group (AECG) criteria for pSS,12 but because the study was performed prior to the publication of the EULAR primary Sjögren's syndrome disease activity index (ESSDAI)13 in 2010, validated disease activity measures were not available. Untreated hypothyreosis was an exclusion criterion. Patients were recruited from Norway (n=227) (Bergen (n=135), Stavanger (n=92)), Sweden (n=140) (Malmö (n=82), Uppsala (n=58)), UK (n=128) (UK Primary Sjögren’s Syndrome Registry) and USA (n=195) (Oklahoma Medical Research Foundation, OMRF). After genotyping quality control (QC) 682 patients remained for further analyses (table 1).
Supplemental material
Clinical characteristics of patients with primary Sjögren’s syndrome included in the association analyses after genotyping quality control
Measures of fatigue
The patients included in the study were evaluated for fatigue with two instruments; the fatigue Visual Analogue Scale (fVAS; all patients from Norway, UK, USA, Uppsala, and n=24 patients from Malmö) and the fatigue item of the EULAR Sjögren’s Syndrome Patient Reported Index (ESSPRI; n=58 patients from Malmö).14 15 The fVAS is a 100 mm horizontal line with vertical anchors. The Norwegian, Swedish and UK edition queried about fatigue during the last week using the wording ‘No fatigue’ at the left anchor (0 mm) and ‘Fatigue as bad as it can be’ at the right anchor (100 mm). The USA fVAS edition queried about fatigue during the last 3 days with the question ‘How would you rate your energy level?’ and the wordings ‘Plenty of energy’ and ‘No energy’ at the left and right anchor, respectively. The ESSPRI fatigue item is a Likert scale (0–10) with the question ‘How severe has your fatigue been the last two weeks?’. Patient ESSPRI fatigue scores were multiplied by 10 to achieve the same range (0–100) as the fVAS. Both instruments were assessed simultaneously in 58 patients showing high correlation (Spearman’s r=0.87, p<0.0001) (online supplemental figure S2).
Genotyping, QC and imputation
The Swedish and Norwegian cohorts were genotyped using the Illumina OmniExpressExome array at the SNP&SEQ Technology Platform, National Genomics Infrastructure, Science for Life Laboratory, Uppsala, Sweden. The UK and USA cohorts were genotyped on the Illumina Omni1-Quad array at the Clinical Genomics Core of the OMRF as previously described.11 Genotype calling was performed using GenomeStudio (Illumina). QC was carried out within the PLINK V.1.9 software framework.16 Only probes present on both genotyping arrays were included in the analysis. Variants with a genotype call rate <95%, Hardy-Weinberg equilibrium p<1×10−4 and/or minor allele frequency (MAF) <5% were excluded. Samples with <97% call rate or excessively increased heterozygosity were excluded, and only samples from unrelated individuals were kept. Principal component analysis (PCA) was performed using Eigensoft (V.6.0.2) to account for population stratification. First, study samples were projected to a combined reference panel (1000 Genomes and Human Genome Diversity Project)17 18 and only samples clustering together with the European population were kept (online supplemental figure S3A). Second, to increase cohort homogeneity, an unsupervised PCA was performed where samples <5 SD from the corresponding cohort mean in the first three dimensions were kept (online supplemental figure S3B). After QC, 682 samples and 451 031 genetic markers were available for imputation.
Prephasing using SHAPEIT2 and genotype imputation using IMPUTE2 with the 1000 Genomes phase 3 integrated haplotypes as reference panel were performed.19 After QC (IMPUTE2 info score ≥0.5 and MAF ≥5%) a total of 4 966 157 markers were available for GWAS.
Statistical and bioinformatics analysis
The combined genotyped and imputed dataset was used in all analyses. First, association analyses in each of the four cohorts were performed separately using a linear regression model as implemented in SNPTEST V.2.5.2 with fatigue as continuous variable (0–100), and including sex and the five first principal components as covariates. Results are presented with beta-value and SE referring to the regression coefficient of the association analysis. Beta-values represent the effect size of the minor allele on the level of fatigue as determined by fVAS (mm). Then meta-analysis of the Norwegian and Swedish cohorts, and the UK and USA cohorts, respectively, was performed in PLINK V.1.9 using a fixed effect weighted z-score approach. Finally, a meta-analysis including all four cohorts was conducted. Genome-wide significance (GWS) was defined as p<5×10−8, and suggestive significance as p<1×10−5. Results were plotted using R Bioconductor (https://bioconductor.org/) and LocusZoom (http://locuszoom.org/). For annotation and functional characterisation HaploReg V.4.1 (http://pubs.broadinstitute.org), Human Protein Atlas (http://proteinatlas.org/) and blood expression quantitative trait loci (eQTL) Browser (http://genenetwork.nl/bloodeqtlbrowser/) were queried.
Receptor transporter protein 4 gene and protein expression analyses
Transcriptome data of peripheral CD19+ B cells were available from 16 pSS patients (Uppsala, Sweden) and 20 matched healthy controls.20 As previously described, mRNA expression was analysed within the Cufflinks/Cuffdiff pipeline, and type I interferon (IFN) scores were calculated based on mRNA expression levels of IFI35, IFITM1, IRF7, MX1 and STAT1.21 Correlations were analysed using Spearman’s rank test, and continuous variables using Mann-Whitney U test applying p<0.05 as significance cut-off. Receptor transporter protein 4 (RTP4) concentration in plasma was measured by ELISA in 92 pSS patients (Stavanger, Norway). All samples were analysed in duplicate and ELISA plates were read on a Synergy H1 microplate reader. Linear regression was performed with fVAS as the dependent variable and RTP4 protein level as the constant.
Results
Fatigue
All four cohorts reported a high level of fatigue as defined as mean fVAS >50. However, fatigue scores differed between the cohorts with significantly lower fVAS in the UK and USA cohorts compared with the Swedish and Norwegian cohorts (table 1, figure 1 and online supplemental figure S4). Fatigue scores did not correlate significantly with age (rs=−0.06, p=0.12) (online supplemental figure S5) or haemoglobin-levels (tested in the Norwegian and Swedish cohort only, results not shown) and did not differ significantly between patients positive for Sjögren’s syndrome-related antigen A (SSA) autoantibodies compared with patients negative for SSA (tested in the Norwegian and Swedish cohorts, p=0.34) (online supplemental table S1).
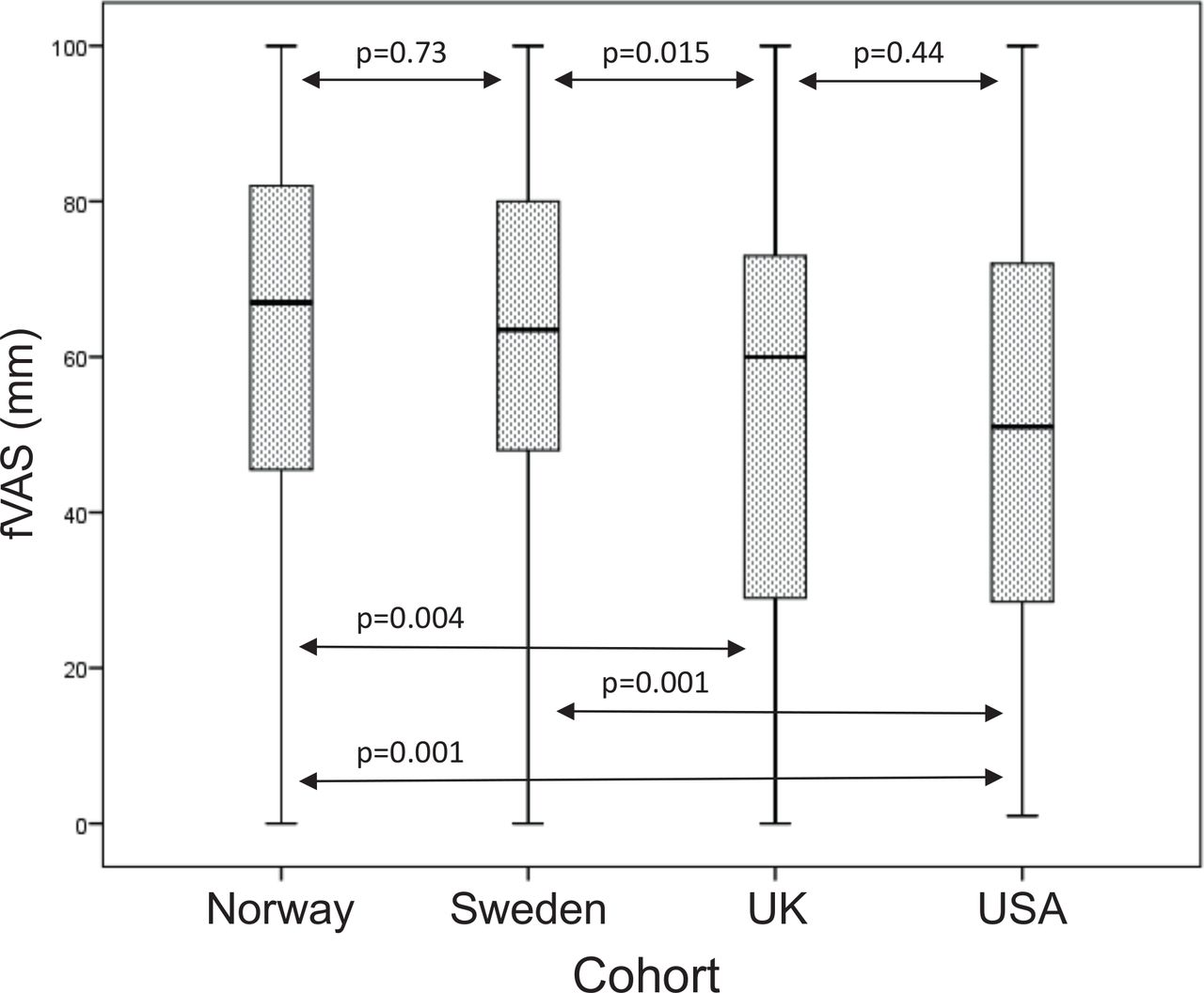
Boxplot of fVAS scores (in mm) in the Norwegian, Swedish, UK and USA cohorts of patients with primary Sjögren’s syndrome. Boxes represent median and IQR, whiskers indicate total range with p<0.05 defining significance. fVAS, fatigue Visual Analogue Scale.
Genome-wide association analysis
Separate analyses in each of the four cohorts
To identify genetic determinants of fatigue in pSS, we used the genome-wide imputed dataset of 4 966 157 autosomal genetic markers (MAF ≥5%). First, we performed association analyses in each of the four cohorts separately (Norway, Sweden, UK, USA). In the Norwegian cohort this analysis revealed 45 variants at six independent loci reaching suggestive significance (p<1×10−5), with the most significant signal for an intronic single nucleotide polymorphism (SNP) at the Chromodomain helicase DNA binding protein 6 gene (rs1969700, p=8.5×10−7, beta (SE)=12.89 (2.55)) (online supplemental table S2). The analysis in the Swedish cohort found 95 variants (p<1×10−5) at eight independent loci. One intronic variant at the Rho GTPase activating protein 26 (ARHGAP26) gene exceeded GWS (rs10039856, p=9.4×10−9, beta (SE)=−32.84 (5.29)) (online supplemental table S3). In addition, strong associations were observed for variants upstream of the RTP4 gene and the Mannan binding lectin serine peptidase 1 (MASP1) gene (top variant rs60344347, p=1.1×10-6, Beta (SE)=−14.96 (2.94)).
Supplemental material
Supplemental material
Association analysis of the UK cohort found 88 variants at twelve independent loci (p<1×10−5), with the top SNP at tumour suppressor candidate 3 gene (rs12679528, p=1.2×10−7, Beta (SE)=17.95 (3.21)) (online supplemental table S4). The separate analysis in the US cohort identified 25 suggestive variants at eight independent loci, with the top signal at the Xin actin binding repeat containing 2 gene (rs7605830, p=2.6×10−6, beta (SE)=−28.18 (5.76)) (online supplemental table S5).
Supplemental material
Supplemental material
Meta-analyses
Since mean and distribution of fatigue scores were similar in Norway and Sweden with significantly higher fatigue than in the UK and US cohorts, we hypothesised that patients from the two Scandinavian countries may share a common genetic predisposition to fatigue (table 1, figure 1, online supplemental figure S4). We, therefore, performed separate meta-analyses of Norway/Sweden and UK/USA, respectively. The meta-analysis in the Norwegian and Swedish cohorts revealed five polymorphisms (one insertion/deletion (indel) and four SNPs) within the same linkage disequilibrium (LD) block (R2 between 0.94–1.0 to top variant rs60344347) located between RTP4 and MASP1 on chromosome 3 that exceeded GWS (figure 2A,B). The minor allele of these variants was associated with less fatigue with a regression coefficient of the top variant rs60344347 (p=3.9×10−8) of beta (SE)=−12.5 (3.08) (table 2). This implicates that compared with an individual being homozygote for the minor allele, on average a heterozygote individual will have a 12.5 mm higher fVAS score, and correspondingly, an individual homozygote for the major allele (risk allele) has a 25 mm higher fVAS score (table 2, online supplemental figure S6). Suggestive significance was exceeded by three additional SNPs within the RTP4/MASP1 region and 45 variants located at twelve other independent loci (figure 2A,B, online supplemental table S6). Of these, SNPs at endoplasmic reticulum to nucleus signalling 1 (ERN1) with top variant rs75160892 (p=9.1×10−7) were the second most significantly associated region after RTP4/MASP1.
Supplemental material

GWAS meta-analysis results of the Scandinavian cohorts. (A) Manhattan plot illustrating the –log 10 p value of all genetic variants analysed in the GWAS meta-analysis of the Norwegian and Swedish cohorts against their chromosomal position. The red horizontal line represents the genome-wide significance threshold (p<5×10−8), the blue line represents the suggestive significance threshold of p<1×10−5. (B) LD link plot with the association p-values on the –log 10 scale of all analysed variants in a 150 kb window around the top associated variant (indel rs60344347) identified in the meta-analysis of the Norwegian and Swedish cohorts plotted against their chromosomal position using the top biallelic SNP (rs7611640) as query variant (indicated in blue) for the regional LD structure in the European reference population, where light red means low LD and dark red corresponds to full LD (R2=f) with the query variant. GWAS, genome-wide association study; LD, linkage disequilibrium; RTP4, receptor transporter protein 4
Genetic variants associated with the level of fatigue in patients with primary Sjögren’s syndrome exceeding genome-wide significance in the meta-analysis of the Norwegian and Swedish cohorts
The meta-analysis of the UK and USA cohorts found no associations exceeding GWS. However, 34 variants at six loci reached suggestive significance, including the PR/SET domain 1 (PRDM1) gene (top SNP rs12175002, p=1.0×10−6) (online supplemental table S7). No association was found between fatigue and polymorphisms in the RTP4/MASP1 locus in the UK and USA cohorts (online supplemental table S8). Finally, we performed a meta-analysis including all four cohorts and observed two suggestive associations with fatigue; an intronic variant at the LIM homeobox 1 locus (LHX1/LHX1-DT) on chromosome 17 (rs10048170, p=9.6×10−7) and at the LOC102723654 on chromosome 5 (rs4509075, p=3.0×10−6) (online supplemental table S9).
Supplemental material
Supplemental material
Supplemental material
RTP4 gene expression and protein expression
Several genetic variants exceeding GWA were identified at the RTP4/MASP1 locus in the meta-analysis of the Scandinavian cohorts. We, therefore, aimed to further characterise potential functions of RTP4/MASP1 and the fatigue-associated variants by investigating gene and protein expression data. RTP4 has a low tissue specificity and is widely expressed in various tissues and cell types. MASP1 expression is specifically seen in liver, brain, female tissues and muscular tissues. We queried public databases eQTL effects of the genetic risk variants identified in the Scandinavian pSS cohorts. eQTLs were reported for the fatigue-associated variant rs7611640 and for two further variants within the same LD block (R2=0.79 to rs6797770, and R2=0.78 to rs1518868) and whole blood gene expression levels of RTP4 and MASP1 (table 3).22 The minor allele of the genetic variant rs7611640 was associated with decreased whole blood mRNA expression in both RTP4 (p=1.4×10−10) and MASP1 (p=2.4×10−3).
Cis-Eqtl effects between genetic variants associated with the level of fatigue in PSS and whole blood gene expression levels at the RTP4/MASP1 locus
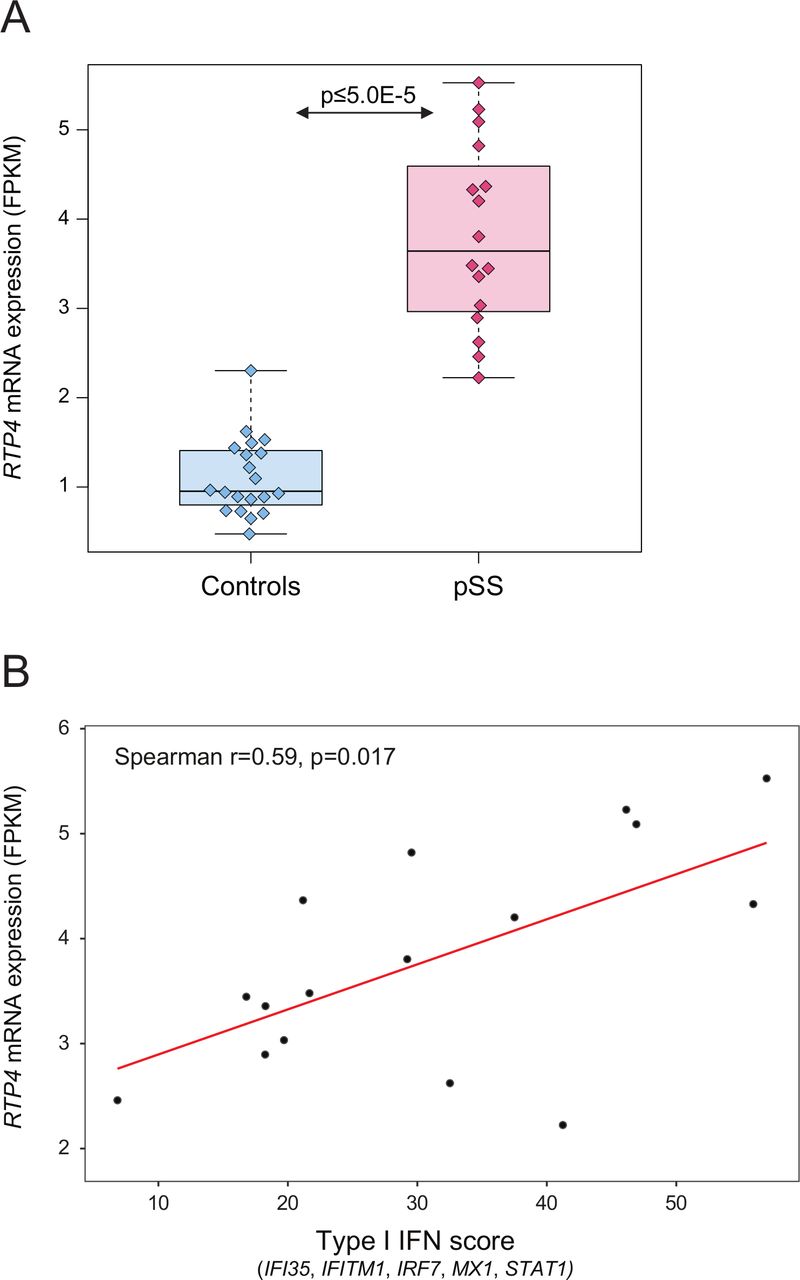
Analysis of gene expression based on RNA-sequencing in pSS patients (n=16) and healthy controls (n=20) from our previous studies revealed upregulation of RTP4 expression in pSS B cells compared with controls (p<5×10−5, fold change=3.4) (figure 3A), while MASP1 expression was generally low in B cells from both patients and controls (data not shown).20 21 In addition, we found a positive correlation of RTP4 gene expression with type I IFN scores in pSS B cells (rs=0.59, p=0.02) (figure 3B). While eQTLs between variants at the RTP4/MASP1 locus and gene expression levels of RTP4 and MASP1 have been reported in public databases (table 3), eQTLs could not be directly studied in our own dataset as the number of patients with both GWAS and RNA-sequencing data was insufficient. Investigation of a potential association between fatigue and RTP4 protein expression in plasma samples from 92 patients with pSS did not reveal a significant result (R2=0.003, p=0.6) (data not shown).

{kind=link}
{kind=link}
{kind=link}
RTP4 mRNA expression (in fragments per kilobase of exon model per million reads mapped (FPKM)) in CD19+ B cells. (A) Boxplot of RTP4 gene expression in n=16 pSS patients and n=20 healthy controls from Uppsala, Sweden. Boxes represent median and IQR, whiskers indicate total range. (B) Spearman’s rank correlation coefficient (r s) between RTP4 gene expression and type I IFN score in n=16 pSS patients with p<0.05 defining significance. FPKM fragments per kilobase of exon per million reads. fVAS, fatigue Visual Analogue scale; IFN, interferon; pSS, primary Sjögren’s syndrome; RTP4, receptor transporter protein 4.
Discussion
The main result of this study is the association between fatigue levels and the RTP4/MASP1 locus on chromosome 3 with five variants exceeding GWS in the meta-analysis of the Norwegian and Swedish pSS cohorts. The major allele of the top variant showed a strong association with elevated levels of fatigue. The associated variants at this locus were in high LD to each other, located upstream of the RTP4 and MASP1 coding regions.
RTP4 encodes the RTP4, a Golgi chaperone involved in the organisation of γ-δ opioid pain receptors. The RTP4 protein enables proper assembly and routing of the receptors to the cell surface and interferes with signalling.23 Pain can be considered a ‘danger signal’ analogue to how pathogens can induce sickness behaviour during infections.5 24 25 Fatigue is a dominant feature in the sickness behaviour response, and the observation of the major allele associated with more fatigue severity may therefore be advantageous. Another aspect of sickness behaviour response is mental depression. Some studies indicate the involvement of RTP4 in severe depression and suicidal behaviour during IFN-a treatment of hepatitis C infections.26 27 These results were extended in a mouse model mimicking IFN-a-related depression; concomitant stimulation with IFN-α and a toll-like receptor (TLR) 3 agonist was necessary to achieve upregulation of IFN-inducible genes, of which RTP4 reached maximum levels within 24 hours.28 Many patients with pSS or other autoimmune diseases have an ‘IFN signature’ on the gene expression level indicating the activation of this pathway.29 30 eQTLs between the fatigue-associated variants at the RTP4/MASP1 locus and gene expression levels of RTP4 and MASP1 have been reported.22 Both are IFN-induced genes, and the minor allele, that is, the variant associated with less fatigue, showed reduced mRNA expression of RTP4 and MASP1 in whole blood. MASP1 encodes a serine protease important for the lectin pathway of complement activation, again strengthening the link between inflammation and fatigue.31 MASP1 cleaves C2 and gives rise to a number of highly active complement components. Complement factor B has been associated with fatigue in a proteomics study of cerebrospinal fluid (CSF) in pSS.32 To further characterise a possible functional impact of genetic variants in pSS-related fatigue, we assessed gene and protein expression levels of RTP4 in pSS patients. Increased mRNA expression of RTP4 in patients compared with controls was found in peripheral B cells, supporting the hypothesis of a putative functional role for RTP4 in fatigue. However, no correlation between fatigue severity and RTP4 protein expression in plasma was observed. It is unknown whether RTP4 protein expression in plasma reflects intracellular and CSF RTP4 levels and activity, and whether complex regulatory mechanisms are involved. Additional studies to further dissect the functional molecular impact of RTP4 and MASP1 in pSS-related fatigue are therefore needed.
Further, the meta-analysis of the Norwegian and Swedish cohorts identified three SNPs within the RTP4/MASP1 locus and 45 variants at eleven other loci reaching suggestive significance (p<1×10−5). This included ERN1 which encodes the Serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1 (IRE1a), a transmembrane resident endoplasmatic reticulum protein important for sensing cellular stress signals. There is a complex interaction between IRE1a and the stress-inducible variant of HSP90a33 which has been associated to severe fatigue in pSS.34 A possible mechanism for HSP90 signalling of fatigue is binding of HSP90α to TLR4 on microglia inducing fatigue through increased IL-1β production.34 35 The exact role of ERN1 and its possible influence on regulatory pathways for fatigue remain to be elucidated.
While the meta-analysis of the UK and USA cohort did not identify any association exceeding GWS, six loci reached suggestive significance, among others variants at the PRDM1 locus with rs12175002 as the top SNP. PRDM1 encodes a transcription factor expressed by T and B cells involved in downregulation of immune responses and repression on IFN-β gene expression. The function of PDRM1 fits well with the hypothesis that downregulating mechanisms of inflammation may be associated with fatigue. Genetic variants at PRDM1 have previously been associated with Crohn’s disease and systemic lupus erythematosus.36 37
Many animal and human studies have documented the importance of proinflammatory cytokines for generation of fatigue, especially IL-1β, and treatment with IL-1 blocking agents has alleviated fatigue in patients with rheumatoid arthritis, diabetes type 2, pSS as well as in cancer.32 34 38–42 There were no direct associations with SNPs in genes coding for cytokines in our data, but as discussed above the pathways of the immune system are complex and it is difficult to pinpoint one specific cytokine or molecule, rather a number of signalling pathways interact and generate fatigue.32
Several other studies aiming to explore the genetics of fatigue in various conditions have been conducted.43–46 Although the top associated variants differ considerably between studies, the general trend points to association with genes involved in regulation of innate immunity, in line with the sickness behaviour theory.
There are some limitations to this study. The number of cases is relatively small, however, the 682 individuals included after rigorous QC constitute the largest cohort in any reported GWAS of fatigue in inflammatory disease to date. In this study we did not perform any genetic analyses of sub-phenotypes of pSS as we believe this would reduce the power to detect genetic associations. Fatigue is a universal phenomenon observed across a wide range of inflammatory, neurodegenerative, cancer and other diseases, without good evidence that some diseases or conditions have more, or less, fatigue than the others.25 In the current study, 505 patients were SSA positive and 177 were SSA negative, both groups too small to perform GWAS with reasonable power to detect genetic associations specific for one of the subphenotypes. However, comparing fatigue levels in the 367 subjects in the Scandinavian cohorts stratified for SSA/SSB status we found no difference in fatigue levels between SSA positive or negative, nor between SSA/SSB positive or negative patients.
Depression scores were not available for all patients and could not be included as a covariate in the regression analyses. However, in inflammatory conditions, depression and fatigue probably share some signalling pathways, making it potentially erroneous to adjust for depression.47 48
Disease activity measures were not available as the data were collected prior to the publication of ESSDAI.13 We cannot exclude the possibility that correction for disease activity could have had an impact on the results. However, across diseases, influence of disease activity measures on fatigue scores appears to be a phenomenon associated with the use of disease-specific fatigue instruments that comprise elements of disease activity or other disease features, not solely those related to fatigue. This association is seldom observed in studies using generic and uni-dimensional fatigue instruments, like we employed in our study.49 50
The results from the Norwegian/Swedish meta-analysis were not replicated in the UK/USA meta-analysis. The lack of replication may indicate spurious results from the Scandinavian meta-analysis, however, we find discrepancies in use of fatigue instruments and accordingly distribution of fatigue between the different cohorts a more plausible explanation. Variation in measurement procedures and socio-cultural influenced differences in perception of fatigue can act as contributing factors. The fVAS version used in Norway and Sweden has the same wording and was applied in the same manner at all sites. The questionnaire that was used in USA differs both in wording and timeframe (last 3 days vs 1 week), and mean as well as distribution of fatigue levels were different from the other cohorts. The words ‘Fatigue as bad as it can be’ at the right end of the fVAS was used in the Norwegian, Swedish and UK cohorts, while the words ‘No energy’ was used in the USA cohort. It is not possible to estimate what this semantic difference means to the individual patients answering the question, but it is doubtless that it might influence the scoring. In addition, the centres in UK used a similar fVAS as in Norway and Sweden, but sent out the form by mail, while in Norway and Sweden the patients answered when visiting the rheumatology clinic.
In addition, underlying genetic heterogeneity may play a role. However, the MAF of associated variants at the RTP4/MASP1 locus was intermediate in patients from the UK (MAF 0.21) and USA (MAF 0.20) compared with the Norwegian cohort (MAF 0.17) and the Swedish cohort (MAF 0.23). The strengths of the current study are inclusion of patients according to well-defined classification criteria and generic and robust measures of unidimensional fatigue.
In conclusion, the results of this GWAS point to genetic variants that may contribute to fatigue in pSS. The top associated variants, RTP4/MASP1, ERN1 and PRDM1, are located at genes related to relevant functions and phenotypes that fit well into the concept of sickness behaviour with fatigue as a major element.
Supplemental material
Data availability statement
Data are available on reasonable request. Genotype data are available on request from the authors on a collaborative basis.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Regional Ethics Committee West, NorwayID: REK 2010/1455 Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank Pascal Pucholt for excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
KBN and JI-K contributed equally.
Contributors KBN and RO conceived the study. KBN, LR, RO, RKD, SJAJ, ET, TM, RJ and W-FN collected patient samples and clinical data. KBN, JI-K, AA and KB analysed the data. KBN, JI-K and RO wrote the manuscript. All authors critically evaluated the manuscript and accepted the final version. RO is the guarantor for this study.
Funding The study was supported by grants from Western Norway Regional Health Authority (Helse vest) (grants 912043, 911807, 911783), the Swedish Research Council (Dnr 2018-02399, Dnr 2016-01982), King Gustav the V:th 80-year foundation, the Swedish Rheumatism association, the Erik, Karin och Gösta Selanders foundation, Uppsala University, and the Swedish Society of Medicine. The UKPSSR was supported by the Medical Research Council (G0800629) and the British Sjögren’s Syndrome Association. The OMRF Sjogren’s Syndrome Cohort has been supported by the National Institutes of Health [grants R01AR50782 (KS), P50AR0608040 (KS), U19AI 082714 (KS), U01DE017593 (KS, AR), R01DE018209 (KS, AR) and R01AR065953 (CJL)]. Additional funding was obtained from the Phileona Foundation (KS, CJL) and the Oklahoma Medical Research Foundation (KS, CJL, AR).
Disclaimer The contents are the sole responsibility of the authors and do not necessarily represent the official views of the NIH.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.