Article Text

Abstract
Objective Evaluate the effect of upadacitinib on pain outcomes in patients with active psoriatic arthritis (PsA) or ankylosing spondylitis (AS) across 3 randomised trials (SELECT-PsA 1 and 2 for PsA; SELECT-AXIS 1 for AS).
Methods Patients were randomised to upadacitinib 15 mg once daily or placebo (all 3 studies), or adalimumab 40 mg every other week (SELECT-PsA 1 only). Pain outcomes included proportion of patients achieving ≥30%, ≥50% and ≥70% reduction from baseline in patient global assessment of pain and other end points.
Results A higher proportion of patients receiving upadacitinib versus placebo achieved ≥30%, ≥50% and ≥70% reduction in pain end points as early as week 2; these improvements with upadacitinib were generally sustained or increased through year 1 (PsA 1/2 studies: 64%/48%, 58%/42% and 38%/22%, respectively; SELECT-AXIS 1 study: 76%, 72% and 54%). Results were similar with adalimumab in PsA 1 (59%, 49% and 32%). Patients who switched from placebo to upadacitinib 15 mg were able to reach a similar level of improvement as the continuous upadacitinib groups by year 1 (PsA 1/2 studies: 46%–60%, 35%–49% and 15%–34%; AS study: 83%, 72% and 46%). Results were similar with other pain end points.
Conclusion Rapid and sustained improvements in pain outcomes across several end points were consistently shown with upadacitinib over 1 year in patients with active PsA or AS who had either inadequate response to prior non-biologic or biologic disease-modifying antirheumatic drugs (PsA studies) or were biologic-naïve with inadequate response to non-steroidal anti-inflammatory drugs (AS study).
- Arthritis, Psoriatic
- Patient Reported Outcome Measures
- Spondylitis, Ankylosing
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual and trial-level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html. NA.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Upadacitinib, a Janus kinase inhibitor, has demonstrated efficacy and safety in patients with active psoriatic arthritis (PsA) in the phase III SELECT-PsA 1 and 2 studies and in patients with active ankylosing spondylitis (AS) in the randomised phase II/III SELECT-AXIS 1 study.
What does this study add?
This analysis of data from three randomised placebo-controlled clinical trials from patients with active PsA or AS demonstrated consistent rapid, clinically meaningful and sustained benefits on various pain end points (including global pain, peripheral/entheseal pain, back pain and nocturnal back pain) with upadacitinib 15 mg once daily.
Reductions were often achieved as early as week 2, and the reductions in pain were sustained over 1 year.
Patients who switched from placebo to upadacitinib 15 mg once daily were able to reach a level of improvement similar to the continuous upadacitinib groups.
How might this impact on clinical practice or future developments?
These findings support the clinical benefit of upadacitinib 15 mg once daily for the improvement of pain in patients with active PsA and inadequate response to prior non-biologic or biologic disease-modifying antirheumatic drugs and in biologic-naïve patients with active AS with inadequate response to non-steroidal anti-inflammatory drugs.
Introduction
Spondyloarthritis is a group of related chronic inflammatory disorders that includes psoriatic arthritis (PsA) and ankylosing spondylitis (AS).1 The primary goal in treating patients with PsA and AS is to maximise health-related quality of life by controlling symptoms and inflammation, preventing structural damage and normalising function and social participation. A recommended treatment target is achieving remission or low disease activity through regular disease activity assessments and appropriate therapy adjustment.2–6 However, this treatment target is not always associated with corresponding improvement in patient-reported outcomes, such as pain and functional impairment. Pain, including inflammatory back pain, joint pain and peripheral/entheseal pain, is a dominant and debilitating symptom of both PsA and AS and can negatively affect patients’ lives.7–10 Back pain is the hallmark of AS, and up to 70% of patients with PsA are reported to have axial involvement.9 11 12 In patients with AS, back pain is associated with fatigue and work impairment,13 14 and in patients with PsA, axial involvement is associated with higher disease activity and greater quality-of-life impairment.11 Thus, pain management is a priority for patients with PsA and AS and often requires therapeutic intervention.2–4 10 15–17 Currently available analgesics are poorly suited for chronic administration in patients with spondyloarthritis, and novel approaches to manage this important element of the disease are required.
In recent years, Janus kinase (JAK) inhibitors have emerged as a new therapeutic class for the treatment of AS and PsA in clinical studies.18–23 Upadacitinib, a JAK inhibitor engineered for increased selectivity for JAK1 over JAK2, JAK3 and tyrosine kinase 2,24 has demonstrated efficacy and safety in patients with PsA in two phase III studies, SELECT-PsA 1 and 225 26 and in patients with active AS in a randomised phase II/III study, SELECT-AXIS 1.27 28 JAK inhibitors also demonstrated improvement in pain (inflammatory and other types) and physical function in rheumatoid arthritis (RA) studies.29–32
The objective of this analysis was to evaluate the efficacy of upadacitinib on pain based on multiple pain assessments through 1 year in patients with active PsA from the SELECT-PsA 1 and 2 studies and in patients with active AS from SELECT-AXIS 1.
Methods
SELECT-PsA 1 and 2 study design and participants
The primary results of the randomised, placebo-controlled phase III SELECT-PsA 1 and 2 studies (NCT03104400 and NCT03104374, respectively) have been previously published.25 26 Briefly, in SELECT-PsA 1, patients with PsA and prior inadequate response (IR) or intolerance to ≥1 non-biologic disease-modifying antirheumatic drugs (DMARDs) but not biologic DMARDs (non-biologic-IR), were randomised to blinded upadacitinib 15 mg once daily, upadacitinib 30 mg once daily, adalimumab 40 mg every other week or placebo for 24 weeks (online supplemental figure 1A). In SELECT-PsA 2, patients with PsA and prior IR or intolerance to ≥1 biologic DMARDs (biologic-IR) were randomised to upadacitinib 15 mg once daily, upadacitinib 30 mg once daily or placebo for 24 weeks (online supplemental figure 1B). In both studies, patients initially receiving placebo were switched to upadacitinib 15 mg once daily or 30 mg once daily (blinded to dose) at week 24 (switch group); patients initially randomised to upadacitinib continued to receive the same dose of upadacitinib (continuous group). At week 16, patients classified as non-responders could add or modify doses of non-biologic DMARDs, non-steroidal anti-inflammatory drugs (NSAIDs), acetaminophen/paracetamol, low potency opioid medications and/or oral glucocorticoids and/or receive one corticosteroid injection.
Supplemental material
Reported here are results for up to 56 weeks of blinded treatment for patients receiving upadacitinib 15 mg once daily, the dosage approved for the treatment of RA, PsA and AS.33 34
The SELECT-PsA programme enrolled adult patients (≥18 years) who had a clinical diagnosis of PsA with symptom onset ≥6 months before screening visit, active PsA (≥3 swollen and ≥3 tender joints), fulfilment of the Classification Criteria for PsA (CASPAR) and had an active or a historical diagnosis of plaque psoriasis. Both studies permitted patients to continue background therapy with NSAIDs, oral glucocorticoids (equivalent to prednisone ≤10 mg/day) and ≤2 non-biologic DMARDs, although this was not a requirement. Patients with prior exposure to any JAK inhibitor were excluded.
SELECT-AXIS 1 study design and participants
The methods and primary results of the randomised, placebo-controlled phase II/III SELECT-AXIS 1 (NCT03178487) study have been previously published.27 Briefly, patients with active AS were randomised 1:1 to upadacitinib 15 mg once daily or placebo for 14 weeks (period 1), followed by open-label upadacitinib 15 mg once daily during a 90-week extension (period 2; online supplemental figure 2). Starting at week 16, patients who did not achieve at least an Assessment of SpondyloArthritis international Society (ASAS) 20 response at two consecutive visits had the option to add or modify doses of NSAIDs, acetaminophen/paracetamol and low potency opioid medications or modify dose of methotrexate or sulfasalazine. Starting at week 24, patients who still had not achieve at least an ASAS20 response at two consecutive visits were discontinued from study drug treatment. Reported here are interim data to up week 64.
The study enrolled adult patients (≥18 years) with a clinical diagnosis of AS who met the modified New York criteria; had active AS, defined as Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and patient assessment of back pain ≥4 (on a numeric rating scale (NRS) 0–10) at screening and baseline; were biologic DMARD-naïve; and had an IR to ≥2 NSAIDs or intolerance to or contraindication for NSAIDs.
Patients receiving concomitant conventional synthetic DMARDs or oral glucocorticoids, NSAIDs and analgesics were eligible. Patients with prior exposure to JAK inhibitors were excluded.
End points
In SELECT-PsA 1 and 2 and SELECT-AXIS 1, clinically meaningful improvements in pain were assessed post hoc as the proportions of patients achieving ≥30%, ≥50% and ≥70% reduction from baseline in patient global assessment of pain (based on “How much pain have you had because of your condition during the last week?) NRS score (0–10).35 36 In addition, the proportions of patients achieving a minimal clinically important difference (MCID; defined as ≥1-point reduction or ≥15% reduction from baseline on a 0–10 NRS) and much better improvement (MBI; defined as ≥2-point reduction and ≥33% reduction from baseline on a 0–10 NRS) in patient global assessment of pain were assessed.35
The following additional end points were assessed in all three studies (referring to the last 7 days): changes from baseline in patient global assessment of pain NRS (0–10) at all time points (prespecified in all three studies), and BASDAI question 2 (neck/back/hip pain; based on “How would you describe the overall level of AS neck, back, or hip pain you have had?”) on NRS (0–10; prespecified in SELECT-AXIS 1), and BASDAI question 3 (peripheral pain/swelling; based on “How would you describe the overall level of pain/swelling in joints other than neck, back or hips you have had?”) on NRS (0–10; prespecified in SELECT-AXIS 1).
In SELECT-PsA 1 and 2 studies, change from baseline in the 36-item Short Form Survey (SF-36) bodily pain domain (raw score range 0–100 with higher scores indicating less pain) was also assessed.
In SELECT-AXIS 1, additional pain end points included the proportions of patients achieving ≥30%, ≥50% and ≥70% reductions from baseline; MCID and MBI in patient assessment of back pain (based on “What is the amount of back pain that you experienced at any time during the last week?”) NRS score (0–10); change from baseline in patient assessment of back pain NRS (0–10); and change from baseline in patient assessment of nocturnal back pain NRS (0–10; based on “What is the amount of back pain at night that you experienced during the last week?”).
Additional post hoc analyses in all three studies included time to ≥30%, ≥50% and ≥70% improvement in patient global assessment of pain and percentage of patients achieving patient global assessment of pain ≤1 NRS and ≤2 NRS over time.
Statistical analyses
In SELECT-PsA 1 and 2, the Cochran-Mantel-Haenszel test adjusting for the stratification factor of current non-biologic DMARD use (yes/no) was used to compare treatments for binary end points; non-responder imputation was used for missing data handling (rescued patients were considered non-responders). For continuous end points, analyses were conducted using the mixed-effects model repeated measures analysis, which included the fixed effects of treatment, visit, treatment-by-visit interaction, the stratification factor of current non-biologic DMARD use (yes/no) and the continuous fixed covariate of baseline measurement. An unstructured variance-covariance matrix was used.
In SELECT-AXIS 1, the Cochran-Mantel-Haenszel test adjusting for the stratification factor of screening high-sensitivity C reactive protein (CRP) level (≤upper limit of normal (ULN) or >ULN) was used to compare treatments for binary end points; non-responder imputation was used for handling missing data. For continuous end points, analyses were also conducted using the mixed-effects model repeated measures analysis, which included the fixed effects of treatment, visit, treatment-by-visit interaction, the stratification factor of screening high-sensitivity CRP level (≤ULN or >ULN) and the continuous fixed covariate of baseline measurement. An unstructured variance-covariance matrix was used in the mixed-effects model repeated measures as well. The median time needed for patients to achieve the thresholds (≥30%, ≥50%, ≥70% improvement in pain) was assessed using the cumulative incidence estimate with competing risks, which included rescue or discontinuation due to lack of efficacy before reaching the thresholds.
The statistical significance defined as p<0.05 was exploratory in nature. All p values are nominal.
Results
In SELECT-PsA 1, 429 patients were randomised to the upadacitinib 15 mg once daily group, 429 patients to the adalimumab 40 mg every other week group and 211 patients to the placebo-to-upadacitinib 15 mg once daily (switch) group (online supplemental figure 1A). Of these, 352 (84.2%), 353 (82.3%) and 172 (81.5%), respectively, completed 56 weeks. In SELECT-PsA 2, 211 patients were randomised to continuous upadacitinib 15 mg once daily and 106 to placebo-to-upadacitinib 15 mg once daily, with 167 (79.1%) and 69 (65.1%), respectively, completing 56 weeks (online supplemental figure 1B). Mean duration since PsA diagnosis varied from 5.9 to 10.3 years, and mean patient global assessment of pain scores were between 6.0 and 6.6 in SELECT-PsA 1 and 2, respectively, at baseline (table 1).
Baseline demographics and disease characteristics in the SELECT-PsA 1, SELECT-PsA 2 and SELECT-AXIS 1 studies
In SELECT-AXIS 1, 93 patients were randomised to upadacitinib 15 mg once daily and 94 patients to placebo (online supplemental figure 2). Of these, 78 (83.9%) in the continuous upadacitinib 15 mg once daily group and 82 (87.2%) in the placebo-to-upadacitinib 15 mg once daily (switch) group completed 64 weeks of treatment. At baseline, mean time since AS diagnosis was 6.0 and 7.8 years, mean patient assessment of pain scores were 6.9 and 6.8 and mean patient assessment of back pain scores were 6.7 and 6.8 in the placebo-to-upadacitinib and upadacitinib groups, respectively (table 1).
Pain end points in psoriatic arthritis
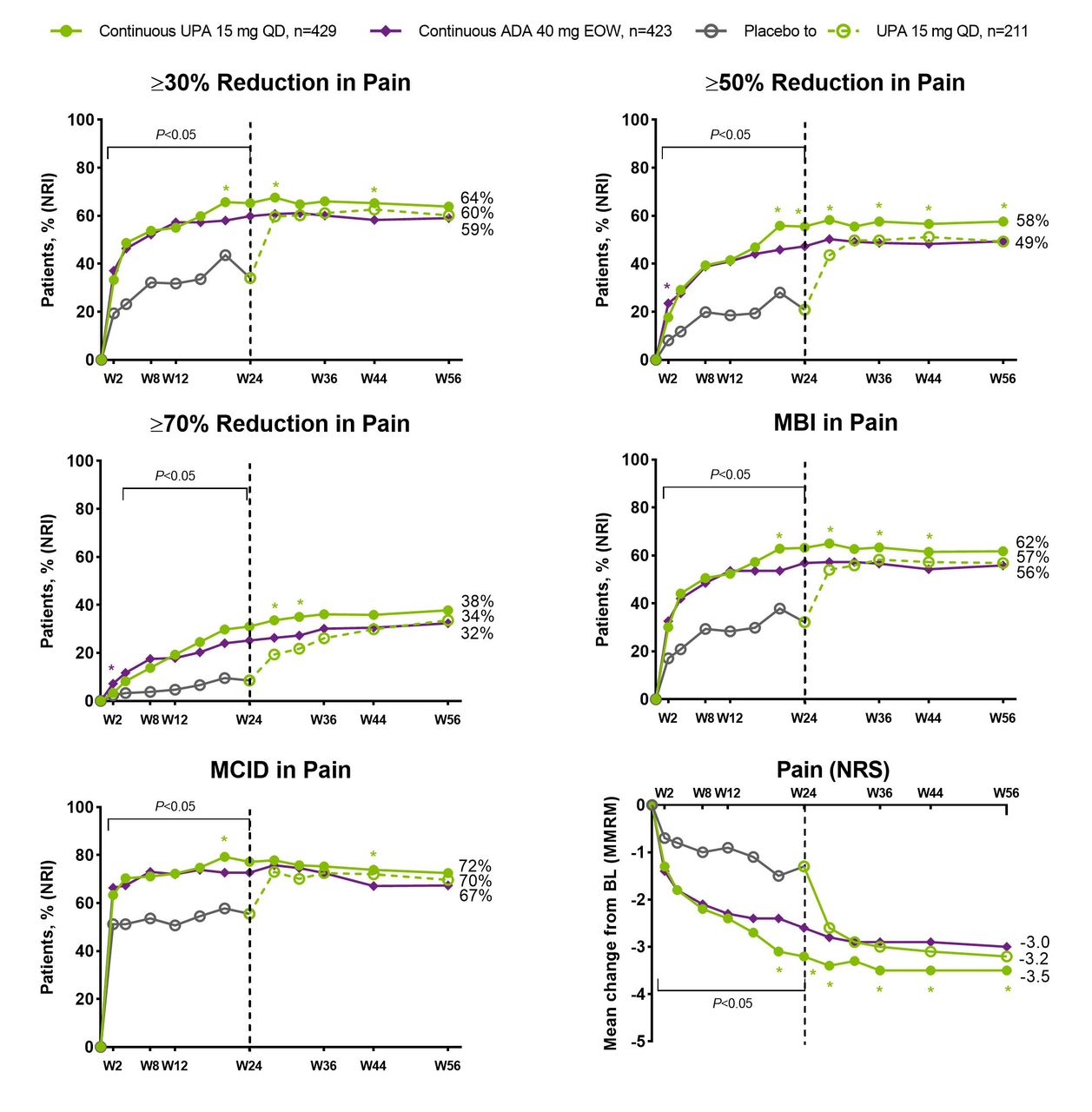
In both PsA studies, significantly higher (nominal p values) proportions of patients receiving upadacitinib 15 mg once daily versus placebo achieved clinically meaningful improvements in patient global assessment of pain, including ≥30%, ≥50% and ≥70% reductions in pain and achievement of MCID or MBI in pain, as early as week 2 for most end points (figure 1 (non-biologic-IR) and figure 2 (biologic-IR)). Of note, approximately two-thirds of patients receiving upadacitinib 15 mg once daily achieved MCID in patient global assessment of pain at week 2 and the response was generally sustained thereafter through week 56 (figures 1 and 2). Median time to ≥30%, ≥50%, ≥70% improvement in pain was significantly (nominal p values) shorter with upadacitinib 15 mg once daily versus placebo in both studies (online supplemental table 1).
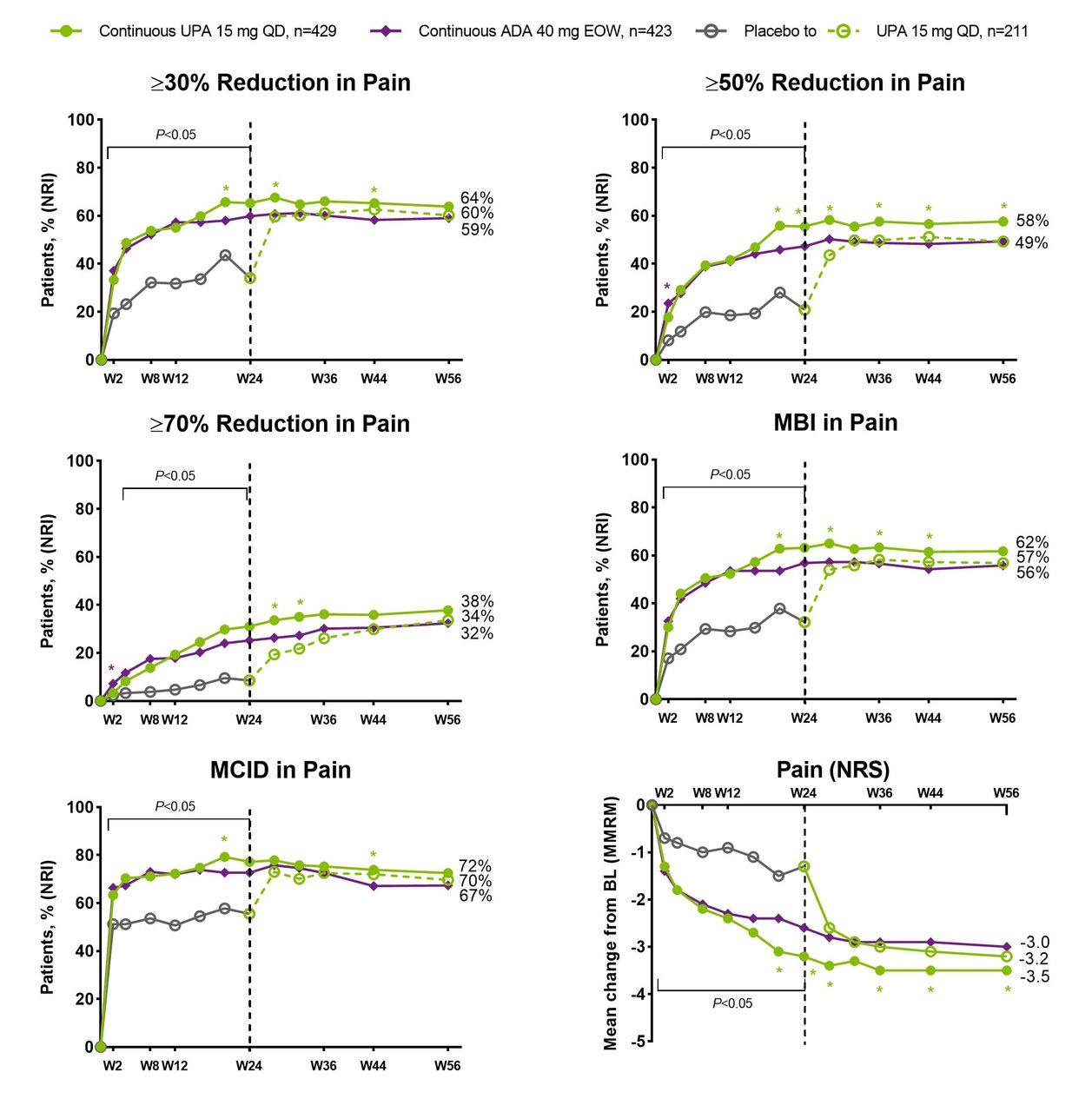
SELECT-PsA 1: percentage of patients achieving ≥30%, ≥50%, ≥70% reduction, MBI or MCID in pain and change from baseline in patient’s global assessment of pain over 56 weeks. ADA, adalimumab; DMARD, disease-modifying antirheumatic drug; EOW, end of week; MBI, much better improvement (≥2-point reduction and ≥33% reduction from baseline on a 0–10 NRS); MCID, minimal clinically important difference (≥1-point reduction or ≥15% reduction from baseline on a 0–10 NRS); MMRM, mixed-effects model for repeated measurements; NRI, non-responder imputation; NRS, numeric rating scale; PBO, placebo; QD, once daily; UPA, upadacitinib; W, week. N’s for NRI analysis; nominal p value for a binary end point was constructed using the Cochran-Mantel-Haenszel test adjusting for the main stratification factor of current non-biologic DMARD use (yes/no). Green asterisks: statistically significant at 0.05 level for continuous UPA 15 mg versus continuous ADA; purple asterisk: statistically significant at 0.05 level for continuous ADA versus continuous UPA 15 mg. Nominal p value above/below the bracket: UPA 15 mg versus overall PBO group statistically significant at 0.05 level at each time point up to week 24, except for week 2 for ≥70% reduction in pain. Dashed line: all patients randomised to PBO received blinded UPA starting from week 24.

SELECT-PsA 2: percentage of patients achieving ≥30%, 50%, 70% reduction, MCID or MBI in patient’s global assessment of pain and change from baseline in patient’s global assessment of pain over 56 weeks. DMARD, disease-modifying antirheumatic drug; MBI, much better improvement (≥2-point reduction and ≥33% reduction from baseline on a 0–10 NRS); MCID, minimal clinically important difference (≥1-point reduction or ≥15% reduction from baseline on a 0–10 NRS); MMRM, mixed-effects model for repeated measurements; NRI, non-responder imputation; NRS, numeric rating scale; PBO, placebo; QD, once daily; UPA, upadacitinib; W, week. N’s for NRI analysis; nominal p value for a binary end point was constructed using the Cochran-Mantel-Haenszel test adjusting for the main stratification factor of current use of non-biologic DMARD (yes/no). Nominal p value above/below the bracket: UPA 15 mg versus overall PBO group at each time point up to week 24 except for week 2 for ≥70% reduction in pain, statistically significant at 0.05 level. Dashed line: all patients randomised to PBO received blinded UPA starting from week 24.
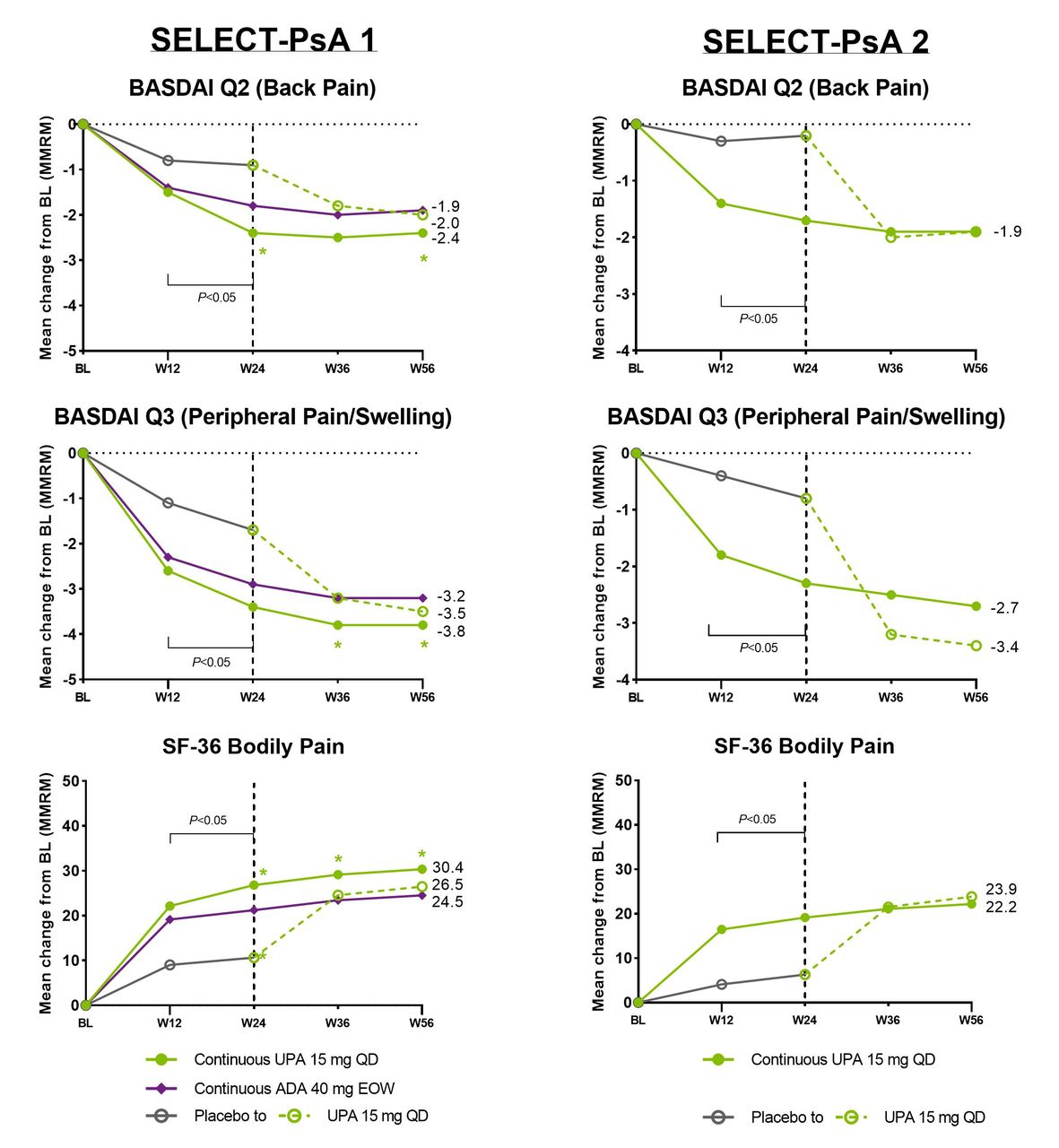
The mean change from baseline in patient global assessment of pain was also significantly greater (nominal p values) for patients receiving upadacitinib 15 mg once daily compared with placebo in both studies from week 2 through week 24. Improvements were generally sustained or increased through week 56. Among patients who switched from placebo to upadacitinib at week 24 in both studies, the magnitude of response observed at week 56 was similar to that observed in the continuous upadacitinib groups (figures 1 and 2). Similar results were observed for BASDAI question 2, BASDAI question 3 and SF-36 bodily pain (figure 3).

Mean change from baseline in BASDAI question 2 and question 3 and SF-36 pain assessments in SELECT-PsA 1 and PsA 2 studies over 56 weeks (MMRM). ADA, adalimumab; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BL, baseline; DMARD, disease-modifying antirheumatic drug; EOW, end of week; MMRM, mixed-effects model for repeated measurements; PBO, placebo; QD, every day; SF-36, 36-item Short Form; UPA, upadacitinib; W, week. In the MMRM, the within-subject dependence was modelled by an unstructured variance-covariance matrix. The fixed effects included the continuous baseline measurement, and treatment, visit, treatment-by-visit interaction and the stratification factor of current non-biologic DMARD use (yes/no) as fixed factors. Green asterisks: statistically significant at 0.05 level for continuous UPA 15 mg versus continuous ADA. Nominal p value above/below the bracket: UPA 15 mg versus overall PBO group at week 12 and week 24; statistically significant at 0.05 level. Dashed line: all patients randomised to PBO received blinded UPA starting from week 24.
Similar proportions of patients receiving upadacitinib 15 mg once daily and adalimumab 40 mg every other week achieved clinically meaningful improvements in pain assessments as early as week 2; differences (nominal p values) favouring upadacitinib 15 mg once daily over adalimumab 40 mg every other week were noted for two or more time points starting at week 20 for all the end points (figures 1 and 3).
Higher proportions of patients receiving upadacitinib 15 mg or adalimumab 40 mg every other week achieved patient global assessment of pain ≤1 NRS and ≤2 NRS versus placebo up to week 24. After week 24, patients who switched from placebo to upadacitinib 15 mg once daily reached a level of pain reduction similar to those initially randomised to upadacitinib in SELECT-PsA 1; however, in SELECT-PsA 2, the response in the placebo-to-upadacitinib group never reached the same level of response over the 56 weeks (online supplemental figure 3).
Pain end points in ankylosing spondylitis
Similar to the results from the PsA studies, in SELECT-AXIS 1, a significantly higher (nominal p value) proportion of patients with AS receiving upadacitinib 15 mg once daily versus placebo achieved clinically meaningful improvements for global assessment of pain, including ≥30% and ≥50% reductions in pain and achievement of MCID and MBI in pain as early as week 2 that were sustained at all time points through week 14; ≥70% reduction in pain was observed at week 4 and sustained thereafter (figure 4). Median time to ≥30%, ≥50%, ≥70% improvement in pain was significantly (nominal p values) shorter with upadacitinib 15 mg once daily versus placebo (online supplemental table 1).
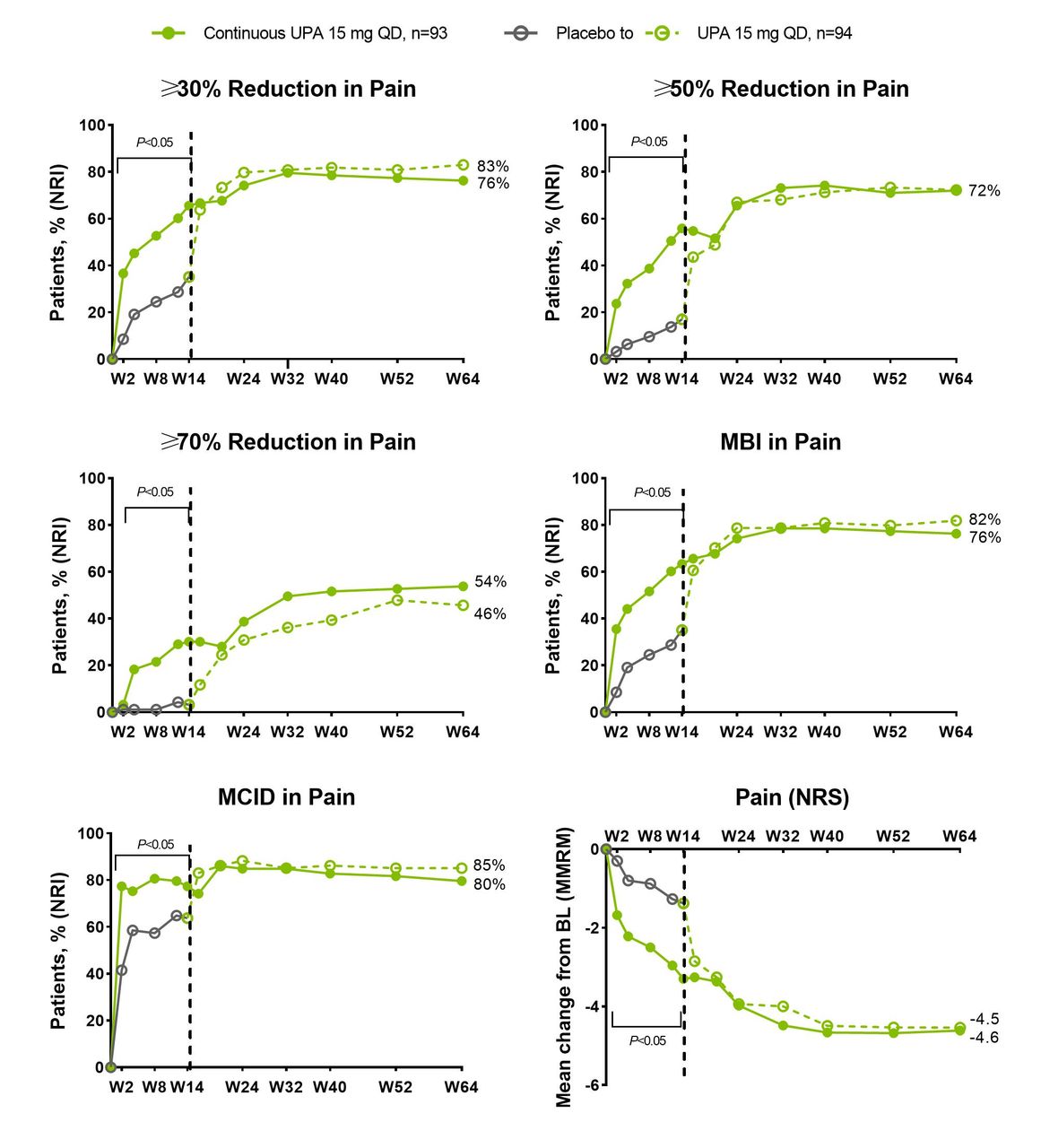
Percentage of patients with ankylosing spondylitis achieving ≥30%, ≥50%, ≥70% reduction from baseline, MBI or MCID in patient’s global assessment of pain through 64 weeks in SELECT-AXIS 1. BL, baseline; CRP, C reactive protein; MBI, much better improvement (≥2-point reduction and ≥33% reduction from baseline on a 0–10 NRS); MCID, minimal clinically important difference (≥1-point reduction or ≥15% reduction from baseline on a 0–10 NRS); MMRM, mixed-effects model for repeated measurements; NRI, non-responder imputation; NRS, numeric rating scale; PBO, placebo; QD, once daily; UPA, upadacitinib; W, week. Dashed line: all patients randomised to placebo received open-label UPA starting from week 14. N’s for NRI analysis; UPA versus PBO comparison was calculated using Cochran-Mantel-Haenszel test adjusting for stratification factor of high-sensitivity CRP level. Nominal p value above/below the bracket: UPA 15 mg versus PBO at each time point up to week 14 except for week 2 for ≥70% reduction in pain; statistically significant at 0.05 level.
Generally, the responses achieved at week 2 increased over time and were sustained through 64 weeks with upadacitinib treatment for ≥30% and ≥50% reduction in patient global assessment of pain and MCID and MBI in pain (achieved by 72%–83% of patients at week 64), whereas 54% of patients achieved ≥70% reduction in pain at week 64 in the continuous upadacitinib 15 mg once daily group (figure 4).
Similarly, the mean change from baseline in patient assessment of pain was significantly greater (nominal p value) with upadacitinib 15 mg once daily compared with placebo at all time points through week 14 (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in pain NRS scores through 64 weeks in SELECT-AXIS 1. BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BL, baseline; CRP, C reactive protein; MMRM, mixed-effects model for repeated measurements; NRS, numeric rating scale; PBO, placebo; QD, once daily; UPA, upadacitinib, W, week. Dashed line: all ankylosing spondylitis patients randomised to PBO received open-label UPA starting from week 14. UPA versus PBO comparison was calculated using Cochran-Mantel-Haenszel test adjusting for stratification factor of high-sensitivity CRP level. Nominal p value below the bracket: UPA 15 mg versus PBO at each time point up to week 14 except for week 2 and 4 for BASDAI question 3; statistically significant at 0.05 level.
Consistent with the patient global assessment of pain end points, the mean change from baseline in patient assessment of back pain, BASDAI question 2 and nocturnal back pain was significantly greater for upadacitinib 15 mg once daily versus placebo as early as week 2 and was maintained at all time points through week 14 (figure 5).
Additionally, when evaluating clinically meaningful improvements in patient assessment of back pain specifically, a significantly higher (nominal p value) proportion of patients receiving upadacitinib 15 mg once daily versus placebo achieved ≥30%, ≥50% and ≥70% reductions and MCID and MBI in patient assessment of back pain; these improvements were also seen as early as week 2 (online supplemental figure 4).
Across all end points, patients who switched from placebo to open-label upadacitinib 15 mg once daily at week 14, generally reached the same level of pain reduction as those initially randomised to upadacitinib (figures 4 and 5, and online supplemental figure 4).
A higher proportion of patients receiving upadacitinib 15 mg achieved pain ≤1 NRS and ≤2 NRS versus placebo up to week 14, after which patients who switched from placebo to upadacitinib 15 mg once daily reached a similar level of pain reduction as those initially randomised to upadacitinib (online supplemental figure 5).
Discussion
This analysis of three randomised clinical trials consistently demonstrated rapid, clinically meaningful and sustained benefits on various pain end points, including global pain, peripheral/entheseal pain, back pain and nocturnal back pain, with upadacitinib 15 mg once daily across patients with active PsA (both non-biologic-IR and biologic-IR) and AS. Significant reductions were often achieved as early as week 2 (the first postbaseline assessment visit in all three studies) and the reductions in pain were sustained over 1 year. Patients who switched from placebo to upadacitinib 15 mg once daily were generally able to reach a similar level of improvement as the continuous upadacitinib groups. In addition, higher proportions of patients with PsA receiving upadacitinib 15 mg once daily versus adalimumab 40 mg every other week achieved improvements in several pain assessments from week 20 onward.
In SELECT-AXIS 1, improvement in back pain was consistent with global assessment of pain, likely due to the nature of AS being a primarily axial disease. Considering that AS is primarily an axial disease and PsA is mainly a peripheral disease, it is notable that upadacitinib also improved peripheral pain (as assessed by BASDAI question 3 in this analysis) in patients with AS and axial pain (as assessed by BASDAI question 2 in this analysis) in patients with PsA over 1 year. Furthermore, a subgroup analysis of the SELECT-PsA 1 and 2 studies demonstrated that upadacitinib 15 mg once daily improved peripheral pain and back pain over 24 weeks compared with placebo.37 Overall, these results suggest that upadacitinib 15 mg once daily consistently improves total pain, axial pain and peripheral pain in patients with PsA, including patients with axial PsA, and AS.
Managing pain is an important part of the treatment of PsA and AS.2 Pain, including inflammatory back pain, is the most commonly reported symptom of AS and PsA, and pain reduction is a priority for patients and treating physicians.10 15 38 Furthermore, pain is associated with reduced quality of life, fatigue and functional and work impairment.13 14 39 Pain at baseline was associated with other disease characteristics, such as swollen and tender joints and spine and joint inflammation, which might have contributed to pain. However, other confounders may also play a role in perception of pain, such as anxiety and depression.40 Currently, data on association of chronic structural damage and pain in patients with AS and PsA are lacking and because structural damage was not uniformly assessed in our studies, no conclusion can be drawn on this particular topic. In RA, MRI-assessed synovitis were associated with pain at 1 or 5 years, while mixed findings for an association with pain were reported for MRI-assessed bone erosions.41 42
Improvement in pain end points have been shown with biologic DMARDs (such as tumour necrosis factor inhibitors, including adalimumab, and interleukin-17A inhibitors) and, perhaps to an even greater extent, with JAK inhibitors in patients with AS, PsA and RA.19 20 29–31 43–45 Although pain in arthritis is predominantly secondary to inflammation, residual, non-inflammatory pain is also common and thought to be caused by joint damage or peripheral and central sensitisation of pain receptors.46 The JAK/signal transducer and activator of the transcription (STAT) pathway is involved in the production of pro-nociceptive and anti-inflammatory cytokines, which in turn can lead to sensitisation of peripheral nerves and pain.47 48 Inhibition of the JAK/STAT pathway can lead to pain modulation, and some patients report pain relief as soon as 24 hours after administration of JAK inhibitors in patients with RA and PsA.47 48 However, it is not known which particular molecular components of the JAK/STAT pathways have relevance to pain in established PsA and AS, and the role of JAK inhibitors on residual pain, independent of inflammation control, needs more investigation.
Our findings are consistent with earlier findings with JAK inhibitors in patients with RA29–32 and show benefits with upadacitinib 15 mg once daily over a broad range of pain end points and clinically meaningful thresholds as early as week 2 (the first time point assessed after baseline) in both AS and PsA populations. The magnitude of pain responses was also consistent between PsA and AS, as well as with RA pain data. Data from three randomised RA studies demonstrated significant improvements in pain with upadacitinib 15 mg once daily versus placebo or methotrexate over 24 weeks.49 Furthermore, upadacitinib 15 mg once daily was also superior to placebo and adalimumab 40 mg every other week for reduction in pain at week 12 in the SELECT-COMPARE RA study.50
The results presented here are, to the knowledge of the authors, the most comprehensive pain analyses reported to date in patients with PsA and AS receiving the JAK inhibitor upadacitinib or the tumour necrosis factor inhibitor adalimumab over 1 year. To our knowledge, this is also the first analysis that assessed MCID and MBI of pain across AS and PsA populations.
Limitations of this post hoc analysis include that patients were not randomised according to their pain level at baseline, SELECT-AXIS 1 was a small phase II/III study (the phase III upadacitinib axial spondyloarthritis/AS programme is ongoing; NCT04169373), all analyses were based on subjective patient assessment of pain and the individual questions from BASDAI have not been validated for individual evaluation and thus were exploratory. However, strengths of this analysis included that baseline characteristics (including pain assessments) were generally well balanced across treatment arms in the studies, patients were blinded to treatment at baseline, the PsA studies included large patient populations, the SELECT-PsA 1 study included an active comparator (adalimumab) arm and importantly, the results were consistent across various pain end points and across AS and PsA populations.
Overall, the results presented here support the clinical benefit of upadacitinib for the improvement of pain in patients with active PsA with IR to prior non-biologic or biologic DMARDs and in biologic-naïve patients with AS with IR to NSAIDs.
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual and trial-level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html. NA.
Ethics approval
All patients provided written informed consent, and all three studies were conducted in accordance with International Council for Harmonisation guidelines, applicable regulations and guidelines governing clinical study conduct, and the ethical principles of the Declaration of Helsinki. Each study and all study-related documents were approved by independent ethics committees or institutional review boards.
Acknowledgments
Medical writing support was provided by Maria Hovenden, PhD, and Janet Matsuura, PhD, of ICON (Blue Bell, Pennsylvania, USA) and was funded by AbbVie.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors IBM, PJM, AD, RL, AM and I-HS participated in the design of the study. All authors analysed and interpreted the data. DF and TG conducted the statistical analysis. Maria Hovenden prepared the first draft of the manuscript. All authors reviewed and critically revised drafts of the manuscript and approved the final draft. IBM is the guarantor for this paper.
Funding AbbVie funded these studies and participated in the study designs, research, analyses, data collection, interpretation of data, reviewing and approval of the manuscript.
Competing interests IBM has received research grants, and/or consulting fees from AbbVie, AstraZeneca, Boehringer Ingelheim, Bristol Myers, Celgene, Janssen, Leo, Lilly, Novartis, Pfizer and UCB. AJKO has served as a consultant and/or on advisory boards for AbbVie, BMS, Gilead, Roche, Janssen, Lilly, Novartis, Paradigm, Pfizer and UCB. PJM has received research grants, consulting fees and/or speaker’s fees from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers, Celgene, Galapagos, Genentech, Gilead, GlaxoSmithKline, Janssen, Leo, Lilly, Merck, Novartis, Pfizer, Sun Pharma and UCB. WT has served on advisory boards or as a consultant or received speaker fees from AbbVie, Amgen, Celgene, Janssen, Lilly, MSD, Novartis, Pfizer and UCB. XB has received honoraria and consultancy fees from AbbVie, Amgen, BMS, Chugai, Galapagos, Gilead, Lilly, MSD, Novartis, Pfizer, Roche, Sandoz and UCB. KdV has received grant/research support from Celgene and Galapagos; consultancy fees from Celgene, Eli Lilly, Galapagos, Novartis and UCB; and speaker fees from Celgene, Eli Lilly, Galapagos, Novartis and UCB. LB has received grant/research support from AbbVie, Amgen, BMS, Celgene, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, Sanofi and UCB; received speaker fees from AbbVie, Amgen, BMS, Celgene, Fresenius Kabi, Janssen, Lilly, Merck, Novartis, Pfizer, Sanofi, Teva and UCB; and served as an officer or board member for AbbVie, Amgen, BMS, Celgene, Fresenius Kabi, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, Sanofi and UCB. RL, AM, DF, TG, PZ, CS, KK and I-HS are full-time, salaried employees of AbbVie and may own AbbVie stock or stock options. AD has received research grants, consultancy fees, speaker fees and other support (medical writing support) from Novartis and Pfizer; research grants, consultancy fees and other support (medical writing support) from AbbVie, Eli Lilly and UCB Pharma; research grants and consultancy fees from GlaxoSmithKline; consultancy fees and other support (medical writing support) from Galapagos and Janssen; consultancy fees from Boehringer Ingelheim and Celgene; and other support (medical writing support) from Amgen.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.