Article Text

Abstract
Background 52-week results from C-axSpAnd demonstrated the safety and efficacy of certolizumab pegol (CZP) in patients with active non-radiographic axial spondyloarthritis (nr-axSpA) and objective signs of inflammation (sacroiliitis on MRI and/or elevated C-reactive protein levels). Long-term safety and clinical outcomes, including MRI assessments, are evaluated up to 3 years for CZP-treated patients with nr-axSpA.
Methods C-axSpAnd was a phase 3 study comprising a 1-year double-blind, placebo-controlled period and 2-year open-label safety follow-up extension (SFE). At baseline, 317 patients were randomised 1:1 to placebo or CZP 200 mg every 2 weeks. Patients completing the double-blind phase who enrolled into the SFE received open-label CZP for an additional 104 weeks. Long-term safety and clinical outcomes are reported to Week 156. Continuous outcomes are presented as observed case (OC) and dichotomous outcomes as OC and with non-responder imputation.
Results 243/317 (76.7%) patients entered the SFE, during which 149 (61.3%) experienced ≥1 treatment-emergent adverse event (TEAE); 15 (3.3/100 patient-years) experienced serious TEAEs. Continuous outcome scores (including Ankylosing Spondylitis Disease Activity Score [ASDAS]: 1.8; Bath Ankylosing Spondylitis Disease Activity Index [BASDAI]: 2.7) at Week 52 were maintained at Week 156 (ASDAS: 1.8; BASDAI: 2.6) for the initial CZP-randomised group. Mean SPARCC MRI sacroiliac joint inflammation scores for these patients decreased at Week 52 (baseline: 7.6; Week 52: 1.7), remaining low at Week 156 (2.4).
Conclusions CZP treatment was well tolerated up to 3 years, with no new safety signals versus previous reports. Clinical outcomes achieved after 1 year were sustained to 3 years.
Trial registration number NCT02552212.
- certolizumab pegol
- spondylitis, ankylosing
- tumour necrosis factor inhibitors
Data availability statement
Data are available upon reasonable request. Data from this manuscript may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised IPD and redacted study documents which may include: raw datasets, analysis-ready datasets, study protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password protected portal.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Certolizumab pegol (CZP) was shown to be clinically effective and well tolerated during the 52-week C-axSpAnd study for patients with non-radiographic axial spondyloarthritis (nr-axSpA) and objective signs of inflammation.
What does this study add?
This is the first study exploring the long-term safety and clinical outcomes of patients with nr-axSpA and objective signs of inflammation, treated with CZP for up to 3 years.
Treatment-emergent adverse events were recorded at regular safety assessments in a systematic and consistent manner, providing the opportunity to identify any potential new safety concerns with long-term cumulative CZP exposure in this patient group. No new safety signals were identified during this study.
Clinical outcomes reported at Week 52 were shown to be sustained at Week 156 for patients continuing CZP treatment, with the use of MRI of sacroiliac joints at Weeks 52 and 156 providing objective assessments of inflammation to further support these clinical data.
How might this impact on clinical practice or further developments?
The safety profile and sustained clinical outcomes observed in this 3-year study support long-term CZP treatment as a suitable option for patients with nr-axSpA and objective signs of inflammation.
Introduction
Non-radiographic axial spondyloarthritis (nr-axSpA) is a chronic inflammatory disease affecting the sacroiliac (SI) joints and spine, but without significant sacroiliitis detected on radiographs.1 2 Objective signs of inflammation, such as sacroiliitis on MRI and/or elevated levels of C-reactive protein (CRP) are often observed, and patients with nr-axSpA exhibit comparable clinical characteristics and disease burden to those with radiographic axSpA.1 2 Long-term suppression of inflammation and the prevention of structural damage are important goals for the treatment of nr-axSpA.3 4
Certolizumab pegol (CZP) is an Fc-free, PEGylated tumour necrosis factor inhibitor (TNFi) which has previously demonstrated efficacy in patients across the axSpA spectrum with an acceptable safety profile.5–9 CZP was approved by the European Medicines Agency in 2013 as one of the first TNFi treatments for use in patients with nr-axSpA.10 The phase 3 C-axSpAnd study evaluated the efficacy of CZP versus placebo in a population of patients with nr-axSpA and objective signs of inflammation. The findings from the 52-week placebo-controlled, double-blind period of C-axSpAnd resulted in FDA regulatory approval for CZP as the first biological to be indicated for treatment of patients with nr-axSpA in the USA.11 12
The long-term safety and maintenance of clinical response to TNFi treatment, beyond a 52-week controlled study setting, in patients with nr-axSpA and objective signs of inflammation has not been previously reported. These data are of key importance since treatment over several years is likely to be required in patients with nr-axSpA to maintain disease control.11 This has been highlighted by an increased risk of flare upon treatment withdrawal in patients with nr-axSpA who have achieved sustained remission.13–15
Here, we report safety and clinical outcomes, including MRI assessments, from the 3-year C-axSpAnd study with a focus on the 2-year safety follow-up extension (SFE).
Methods
Study design
C-axSpAnd (NCT02552212) was a 3-year, phase 3 multicentre study investigating the efficacy and safety of CZP in patients with nr-axSpA and objective signs of inflammation (study design shown in online supplemental figure 1).11 Patients were screened from September 2015 and subsequently randomised 1:1 to CZP (400 mg loading dose at weeks 0, 2 and 4, followed by 200 mg every 2 weeks [Q2W]) or placebo, which they received in addition to their current non-biological background medication (NBBM) in a 52 week, parallel-group, double-blind, placebo-controlled period. Patients from both treatment groups could make changes to their NBBM or switch to open-label CZP at any time during the study, although changes before Week 12 were discouraged. Changes to NBBM were subject to the previously published study restrictions and details of concomitant NBBM permitted during the study are provided in online supplemental table 1.11 At Week 52, patients from both initial treatment groups (including those who had switched to open-label CZP) completing the double-blind period and consenting to enter the SFE received open-label CZP 200 mg Q2W (in addition to NBBM) for an additional 104 weeks. Clinical outcomes were assessed at Weeks 52 and 156 only.
Supplemental material
Patients
Inclusion and exclusion criteria for the C-axSpAnd study have been reported previously.11 Eligible patients were ≥18 years of age, with a clinical diagnosis of adult-onset nr-axSpA meeting the Assessment of SpondyloArthritis international Society (ASAS) criteria,16 and not meeting the modified New York classification criteria (as confirmed by centrally read SI joint radiographs), symptom duration for ≥12 months, active disease (Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] ≥4 and spinal pain ≥4), and inadequate response, intolerance, or contraindication to ≥2 non-steroidal anti-inflammatory drugs. Patients were excluded from the study if they had previous exposure to more than one biological treatment (specifically, TNFi), as per the previously published exclusion criteria.11 Patients were also required to have objective signs of inflammation (a centrally read MRI scan showing active sacroiliitis,17 and/or baseline CRP levels above the upper limit of normal [measured in a central laboratory using a threshold of ≥10.0 mg/L]).
Study assessments
The analysis evaluated here includes only those patients who entered the 104-week SFE at Week 52, analysed by the initial treatment groups to which patients were randomised at study baseline. The primary objective of the SFE was to assess the long-term safety of CZP treatment, as measured by the incidence of treatment-emergent adverse events (TEAEs), severe TEAEs (as measured by intensity), subject discontinuations due to TEAEs, TEAEs leading to permanent withdrawal of study medication, drug-related TEAEs, serious TEAEs, previously unreported TEAEs, and deaths.
TEAEs of interest (defined as opportunistic infections, hepatic events, hypersensitivity and anaphylactic events, malignancies including lymphoma, serious cardiovascular events, haematopoietic cytopenia, serious bleeding events and demyelinating-like disorders), frequently reported TEAEs (those reported in ≥5% of patients in any randomisation group), extra musculoskeletal manifestations and Candida infections were also evaluated.
Adverse events were documented throughout the SFE period and regularly assessed during visits to study centres every 12 weeks (starting at Week 64).
Evaluation of long-term changes to prespecified clinical outcomes (assessed at Weeks 52 and 156) for patients treated with CZP was a secondary objective. Continuous clinical outcomes reported are: Ankylosing Spondylitis Disease Activity Score (ASDAS), BASDAI, Bath Ankylosing Spondylitis Functional Index (BASFI), SpondyloArthritis Research Consortium of Canada (SPARCC) MRI SI joint inflammation score,17 Patient Global Assessment of Disease Activity (PGADA), nocturnal spinal pain, total spinal pain and the Ankylosing Spondylitis Quality of Life (ASQoL) questionnaire (with a scale of 0–1).
Dichotomous clinical outcomes reported are ASDAS Major Improvement (ASDAS-MI), ASAS ≥40% improvement (ASAS40) and BASDAI ≥50% improvement (BASDAI50). Post hoc analyses of ASDAS disease states are also reported.
Statistical analysis
Safety and clinical outcome data are reported for patients who entered the SFE at Week 52 and were analysed descriptively by initial randomisation groups (i.e. CZP 200 mg Q2W or placebo). For adverse events of any type, exposure-adjusted incidence rates (EAIRs) per 100 patient years (PY) were calculated for exposure to CZP.
For continuous outcomes, data are reported as observed case (OC). Given the 2-year gap in assessments between Week 52 and Week 156, missing values for continuous outcomes were not imputed as this would involve using available data collected at least 2 years prior to the Week 156 visit for imputation. Dichotomous outcomes were calculated relative to study baseline (Week 0) at Weeks 52 and 156 and are reported as OC and using non-responder imputation (NRI).
Results
Patient disposition and baseline characteristics
Of the 317 patients entering the double-blind period (CZP: n=159; placebo: n=158), 243 (76.7%) consented to enter the SFE at Week 52 (CZP: n=120; placebo: n=123; figure 1). 10/120 (8.3%) patients entering the SFE and initially randomised to receive CZP switched to open-label CZP during the double-blind period. 75/123 (61.0%) patients entering the SFE, and initially randomised to placebo, switched to open-label CZP before Week 52; the remaining 48/123 patients completed the double-blind period on placebo. For the 96/158 patients who switched to open-label CZP during the double-blind period (which includes the 75 who entered the SFE; figure 1), the mean (standard deviation [SD]) time between initial randomisation and switching to open-label CZP was 18.0 (6.4) weeks.
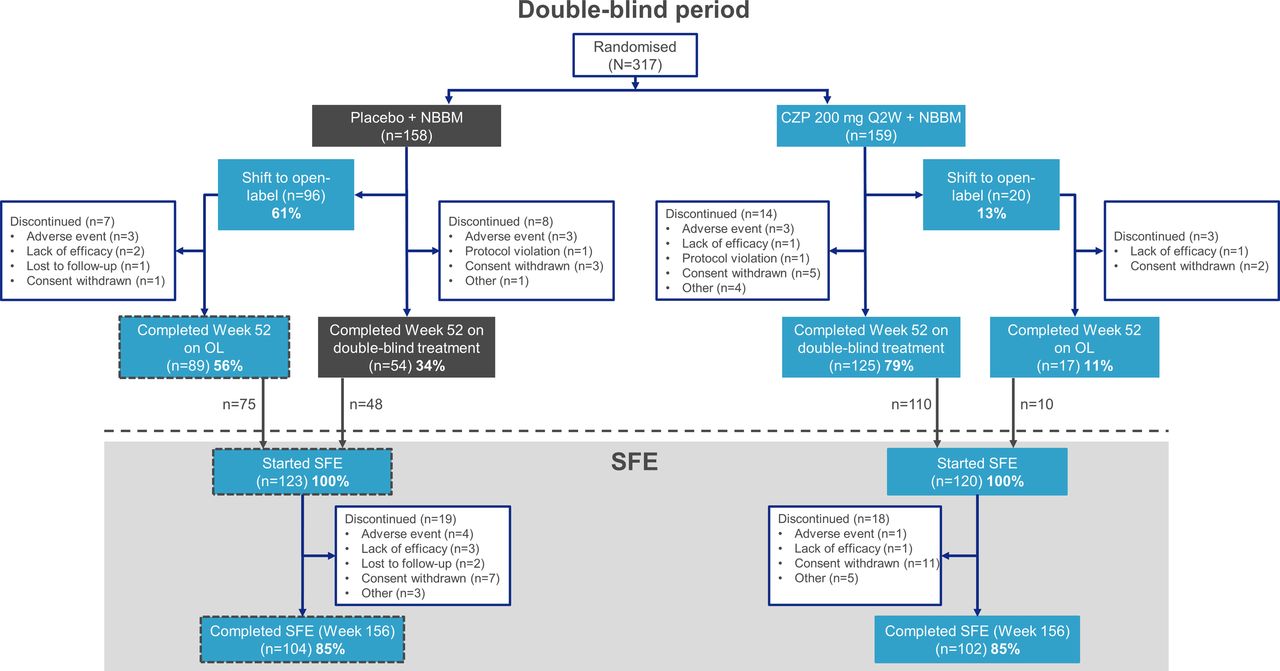
Patient disposition and retention to week 156. Randomised set (N=317). Percentages shown are calculated using the total number of patients starting the specified study period, and within the respective treatment arm, as the denominator. CZP, certolizumab pegol; NBBM, non-biological background medication; OL, open-label; Q2W, every 2 weeks; SFE, safety follow-up extension.
In total, 206/243 (84.8%) patients completed the SFE period up to Week 156, including 102/120 (85.0%) from the initially randomised CZP arm and 104/123 (84.6%) from the initially randomised placebo. 206/317 (65.0%) of patients randomised at baseline completed both the double-blind and SFE periods (102/159 [64.2%]) initially randomised to CZP and 104/158 [65.8%] initially randomised to placebo).
Baseline characteristics, at Week 0, of patients who entered the SFE are reported in table 1. The mean age of patients entering the SFE was 37.1 years, with a mean symptom duration of 7.4 years; 124/243 (51.0%) of patients were male and 209/243 (86.0%) were human leucocyte antigen-B27 positive. During the SFE, 77/243 (31.7%) patients received concomitant conventional synthetic disease-modifying antirheumatic drugs. Baseline characteristics, at Week 0, of patients who completed the double-blind period, but did not enter the SFE, are reported in online supplemental table 2.
Baseline patient demographics and characteristics for patients participating in the safety follow-up extension study
Safety
Total cumulative exposure to CZP during the SFE was 459.8 PY (232.0 PY for those initially randomised to CZP and 227.9 PY for the former placebo arm). The mean (SD) exposure to CZP was 98.7 (20.2) weeks per patient for those entering the SFE (N=243).
In total, 149/243 (61.3%) patients experienced a TEAE in the SFE period giving an EAIR of 57.3 (table 2). Six (2.5%) patients experienced TEAEs leading to the permanent withdrawal of study treatment during the SFE (seven events comprising one case each of hypoacusis, gastroenteritis rotavirus, furuncle, upper respiratory tract infection and erythema multiforme and two cases of tuberculosis infection in patients with a negative QuantiFERON test at Week 0 [no further QuantiFERON tests were carried out during the trial, unless tuberculosis was suspected]), with a combined EAIR of 1.3. The two cases of tuberculosis recorded during the SFE occurred in Russia and the Czech Republic, the former of which has a high background prevalence.18 Neither patient had evidence of latent tuberculosis at baseline. One of these cases was confirmed to be pulmonary tuberculosis which was reported as not resolved at the time of the last visit of this patient in the study, while the other was unspecified and resolved 58 days after onset.
Incidence of TEAEs during the double-blind period and safety follow-up extension
For the SFE period, 36/243 (14.8%) patients were deemed to have TEAEs related to CZP by the study investigators. Serious TEAEs were reported for 15/243 (6.2%) patients during the SFE period with an EAIR of 3.3 (table 3). Of the 20 serious TEAEs reported in 15 patients, four (including the two cases of tuberculosis and one each of chronic tonsilitis and erythema multiforme) were judged to be treatment-related by the study investigators. Incidence of TEAEs for patients receiving additional background medication during the SFE are given in online supplemental table 3.
Serious TEAEs reported during the safety follow-up extension
During the SFE, seven patients were recorded as having uveitis in total, including two cases of uveitis which required hospitalisation as per the clinical practice in the countries in which these cases were reported (Poland and Russia), giving an EAIR of 1.5 (0.4 for the serious TEAEs). Hospitalisation of these patients was not due to the nature of the uveitis recorded; however, as hospitalisation occurred, this required the classification of these cases as serious TEAEs (as per the study protocol). Of the seven patients with uveitis, five had history of the condition. Two patients were recorded as having inflammatory bowel disease (IBD) and one patient was recorded as having psoriasis, giving EAIRs of 0.4 and 0.2, respectively. Of the two patients recorded as having IBD during the SFE, one had no history but reported ongoing ‘irritable bowel syndrome’; the other patient reported prior history of IBD. The patient experiencing psoriasis had no history of the condition.
Opportunistic infections occurred in three patients with an EAIR of 0.7 during the SFE across all patients. These three events included one case of herpes zoster and two of tuberculosis. The latter two patients were withdrawn from study treatment and referred to an appropriate tuberculosis specialist as per the study protocol. One patient experienced a Candida infection (oral candidiasis) during the SFE, with an EAIR of 0.2.
Two patients experienced a TEAE of increased alanine aminotransferase levels (≥3 times the upper limit of normal) during the SFE (accounting for the two reported hepatic events). There were also two hypersensitivity reactions which occurred in a single patient. No malignancies, serious cardiovascular events, haematopoietic cytopenia, serious bleeding events or demyelinating-like disorders were reported during the SFE.
The most frequently reported TEAEs (≥5% of patients in any randomisation group; table 2) during the SFE period were nasopharyngitis (EAIR: 6.0), upper respiratory tract infections (EAIR: 4.8), tonsilitis (EAIR: 2.7) and bronchitis (EAIR: 2.2). No deaths were reported, and no new safety signals were identified during the SFE compared with the previous study report.11
Clinical outcomes
Mean scores for the group of patients initially randomised to receive CZP showed numerical decreases, indicative of clinical improvements, in all reported continuous outcomes at Week 52. The clinical outcomes achieved at Week 52 were maintained to Week 156; however, BASFI scores were observed to further decrease from 2.3 to 1.9 in Weeks 52–156 for this arm (OC data; figure 2).
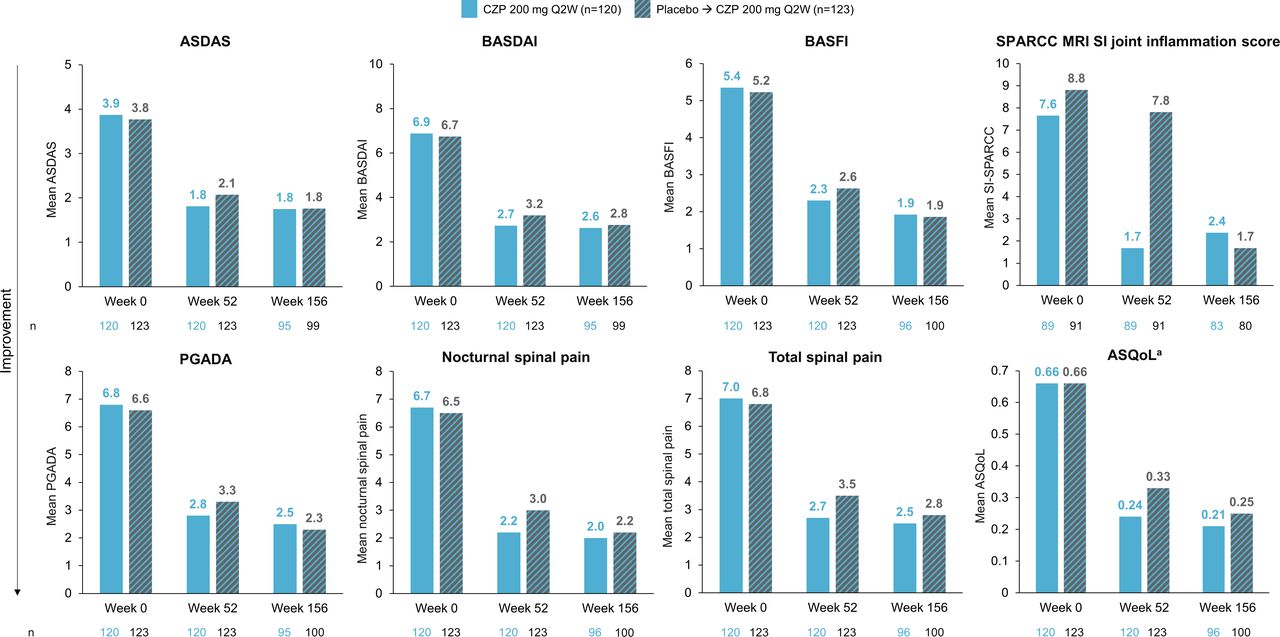
Continuous measures of clinical outcomes reported up to Week 156 (OC). aASQoL measured on a scale of 0–1. Mean values for clinical outcomes at Week 0, 52 and 156 using OC analysis. ASDAS, Ankylosing Spondylitis Disease Activity Score; ASQoL, Ankylosing Spondylitis Quality of Life; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; CZP, certolizumab pegol; OC, observed case; PGADA, Patients Global Assessment of Disease Activity; Q2W, every 2 weeks; SI, sacroiliac; SPARCC, SpondyloArthritis Research Consortium of Canada.
Numerical decreases were observed at Week 52 in all reported continuous clinical measures for the group of patients initially randomised to placebo (including those who received open-label CZP during the double-blind phase). At Week 156, additional decreases in these scores were observed and aligned with those reported for the group of patients randomised to CZP (OC data). ASQoL scores followed this trend and were comparable between groups at Week 156 (CZP-randomised: 0.21; placebo-randomised: 0.25).
Mean SPARCC MRI SI joint inflammation scores (OC) for the CZP-randomised group (baseline: 7.6; Week 52: 1.7) showed a numerical decrease at Week 52; however, the placebo-randomised group did not show any substantial change from Week 0 (8.8) to Week 52 (7.8). At Week 156, the mean SPARCC MRI SI joint inflammation score for the group of patients initially randomised to CZP remained low (2.4) and, for the group initially randomised to placebo, it had decreased to a similar level (1.7).
The percentage of CZP-randomised patients achieving ASAS40 and BASDAI50 showed small decreases at Week 156 when applying NRI; however, OC data for these measures showed that a similar percentage of CZP-randomised and placebo-randomised patients achieved these outcomes at Week 156 (figure 3). ASDAS-MI showed numerical decreases in the percentage of CZP-randomised patients achieving this threshold at Week 156 for both OC and NRI data.
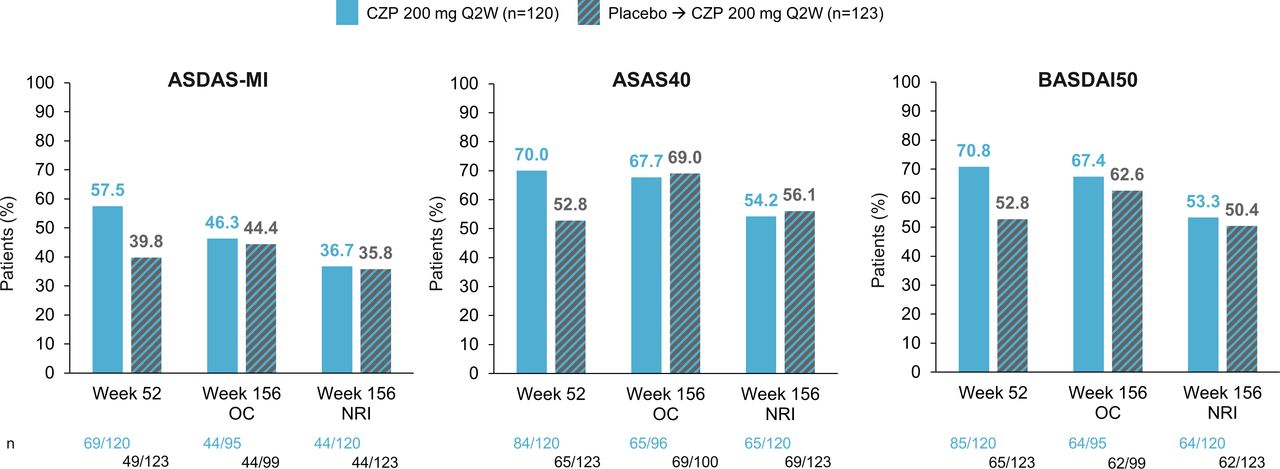
The proportion of patients achieving ASDAS-MI, ASAS40, BASDAI50 (OC and NRI). ASAS40, Assessment of SpondyloArthritis international Society ≥40% improvement; ASDAS: Ankylosing Spondylitis Disease Activity Score; ASDAS-MI, Ankylosing Spondylitis Disease Activity Score Major Improvement (reduction of ≥2 units from baseline); BASDAI50, Bath Ankylosing Spondylitis Disease Activity Index ≥50% improvement; CZP, certolizumab pegol; NRI: non-responder imputation; OC, observed case; Q2W, every 2 weeks.
The proportion of patients with ASDAS low disease activity (LDA; including those with inactive disease [ID]) was higher for the CZP-randomised group (81/120 [67.5%]) than the placebo-randomised group (65/123 [52.8%]) at Week 52. At Week 156, these proportions were similar between the two groups (CZP: 66/95 [69.5%]; placebo: 67/99 [67.7%]).
The proportion of patients with ASDAS-LDA (ASDAS ≥1.3–<2.1) at Week 52 was slightly higher in the CZP-randomised group (40/120 [33.3%]) than placebo-randomised group (31/123 [25.2%]); however, these were similar at Week 156 (CZP: 28/95 [29.5%]; placebo: 30/99 [30.3%]) (OC data; figure 4). At Week 52, ASDAS-ID (ASDAS <1.3) was observed in 41/120 (34.2%) CZP-randomised patients and 34/123 (27.6%) placebo-randomised patients. Both groups showed numerical increases in the percentage of patients achieving ID at Week 156 (CZP: 38/95 [40.0%]; placebo: 37/99 [37.4%]).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The proportion of patients in each ASDAS disease state (OC). ASDAS scores used to assign disease activity were as follows: vHDA: >3.5; HDA: ≥2.1–≤3.5; LDA: ≥1.3–<2.1; ID: <1.3. ASDAS, Ankylosing Spondylitis Disease Activity Score; CZP, certolizumab pegol; HDA, high disease activity; ID, inactive disease; LDA, low disease activity; OC, observed case; Q2W, every 2 weeks; vHDA, very high disease activity.
Discussion
This SFE study allowed the evaluation of the long-term safety and clinical response in patients with nr-axSpA and with objective signs of inflammation, treated with CZP for up to 3 years. Data pertaining to TEAEs were obtained in a systematic and consistent manner, providing the opportunity to identify any potential new safety signals which may have presented with cumulative CZP exposure; the recording of these data in this way represents a strength of this study. An additional strength was in the assessment of stringent measures of disease activity, including ASDAS-ID, ASDAS-MI and ASAS40 responses. The responses to these measures provide evidence to support the sustained effect of CZP treatment at 3 years.
An important consideration when interpreting the findings of the C-axSpAnd SFE is that approximately 15% of the patients who completed the initial 52-week double-blind period did not enter the 2-year SFE. Comparable baseline characteristics were observed between patients entering the SFE and those who did not. Minor differences were reported in the mean symptom duration and time since diagnosis, with both appearing greater for those not entering the SFE. This may present a limitation should these patients be less responsive to treatment; however, this includes a number of patients who were not offered the option to enter the SFE for administrative reasons. Therefore, it is not possible to associate all patient discontinuations with treatment response for the SFE. As such, all of the reported results should be interpreted within this context.
The safety profile was consistent with both that of the 52-week double-blind period and previous clinical trial data for patients with axSpA, treated with CZP.6 7 11 19 20 The most common TEAE reported during the SFE was nasopharyngitis in 26 patients, followed by upper respiratory tract infections in 21 patients. These outcomes are consistent with infections commonly reported as the most frequent TEAE for patients with nr-axSpA treated with TNFi.14 21–23
As with the safety data, the reported clinical outcomes were consistent with previous clinical trial data, including those reported in the RAPID-axSpA and C-OPTIMISE studies.5 6 11 20 Of particular importance, and a further strength of this study, was the use of centrally read MRI to assess SPARCC MRI SI joint inflammation scores at Weeks 52 and 156. These objective assessments of inflammation revealed a numerical decrease in mean score from baseline to Week 52 for the group of patients initially randomised to CZP (from 7.6 to 1.7); at Week 156 this score (2.4) remained substantially lower than that at baseline. Furthermore, the mean SPARCC MRI SI joint inflammation scores for the group of patients initially randomised to placebo were similar at Weeks 0 and 52; however, at Week 156, the mean score had decreased in line with that for the CZP-randomised group. These results indicate the sustained reduction of disease activity, as measured by MRI, with CZP treatment for up to 3 years.
A noteworthy finding was that the percentage of patients achieving ASDAS-MI showed numerical decreases in both OC and NRI data at Week 156 (compared with Week 52), reflecting the disadvantage of a dichotomous response measure. The result is in contrast to the sustained clinical outcomes observed at Week 156 (for the group initially randomised to CZP) and the observed increase in percentage of patients in ASDAS ID and LDA, which is of increased importance as this is a recommended goal for treatment of patients with nr-axSpA.3 4
Although clinical outcomes at Week 156 were sustained from Week 52, no assessments were made in the intervening period, and therefore comparison of these two data points does not allow inference of clinical improvements at additional time points throughout the SFE. Despite this, clinical outcomes at Week 156 do suggest that efficacy after 1 year was sustained at 3 years in patients who continued CZP treatment. Furthermore, most data are primarily presented as OC, which is a limitation of the study. As reporting of observed data may overestimate clinical outcomes, we also reported the more conservative NRI method for dichotomous outcomes, as recommended in the guidelines for the reporting of clinical trial extension studies.24 A further limitation of this study encompasses the unusually long placebo-controlled period of 52 weeks, which may have influenced patients’ decision to participate. In addition, the results may not be generalisable to the general population of patients with nr-axSpA due to the initial inclusion criteria of C-axSpAnd.
The treatment landscape across the axSpA spectrum consists of both TNFi and interleukin (IL)-17 inhibitors, both of which are recommended and have demonstrated efficacy and tolerability for patients with radiographic and nr-axSpA.4 25 CZP is a treatment option which has been evaluated in both patient cohorts and the long-term tolerability and clinical outcomes are demonstrated here for those with nr-axSpA and objective signs of inflammation.
In conclusion, no new safety signals were observed during the 2-year SFE and the safety profile was consistent with previous clinical trial data from CZP in axSpA.5–7 11 20 Furthermore, reduced disease activity, improved patient function and quality of life achieved in the first year of CZP treatment were observed to be sustained at 3 years after a further 2 years of CZP treatment. Overall, our findings support long-term CZP treatment as a suitable option for patients with nr-axSpA and objective signs of inflammation.
Data availability statement
Data are available upon reasonable request. Data from this manuscript may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised IPD and redacted study documents which may include: raw datasets, analysis-ready datasets, study protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password protected portal.
Ethics statements
Patient consent for publication
Ethics approval
The C-axSpAnd study protocol, amendments, and patient informed consent were reviewed by a national, regional or Independent Ethics Committee (IEC) or Institutional Review Board (IRB). This study was conducted in accordance with the current version of the applicable regulatory and International Conference on Harmonisation (ICH)-Good Clinical Practice (GCP) requirements, the ethical principles that have their origin in the principles of the Declaration of Helsinki, and the local laws of the countries involved. Full details of the ethics committee involved are available upon request. All patients provided written informed consent to participate in the study.
Acknowledgments
The authors thank the patients, the investigators and their teams who took part in this study. The authors also acknowledge Luke Green, PhD, from Costello Medical, UK, for medical writing and editorial assistance based on the authors’ input and direction. This study was funded by UCB Pharma.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Substantial contributions to study conception and design: DvdH, LSG, WPM, RL, MR, LB, BH, TK, AD. Substantial contributions to analysis and interpretation of the data: DvdH, LSG, WPM, RL, MR, LB, BH, TK, MK, SEA, AD. Drafting the article or revising it critically for important intellectual content: DvdH, LSG, WPM, RL, MR, LB, BH, TK, MK, SEA, AD. Final approval of the version of the article to be published: DvdH, LSG, WPM, RL, MR, LB, BH, TK, MK, SEA, AD. The corresponding author accepts full responsibility for the work, had access to the data and confirmed the decision to publish.
Funding This study was sponsored by UCB Pharma. This article was based on the original study C-axSpAnd (NCT02552212, ClinicalTrials.gov) sponsored by UCB Pharma. Support for third-party writing assistance for this article, provided by Luke Green, PhD, Costello Medical, UK, was funded by UCB Pharma in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Competing interests DvdH: consultant of AbbVie, Amgen, Astellas, AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Celgene, Cyxone, Daiichi, Eisai, Galapagos, Gilead, GSK, Janssen, Lilly, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda and UCB Pharma. Director of Imaging Rheumatology BV. LSG: research grants from Novartis, Pfizer and UCB Pharma. Consulting fees from AbbVie, GSK, Janssen, Lilly, Novartis, Pfizer and UCB Pharma. WPM: grants from AbbVie, Novartis and Pfizer. Consulting fees from Boehringer Ingelheim, Celgene, Galapagos, Gilead, Janssen, Lilly and UCB Pharma. Honoraria/speakers’ bureau from AbbVie, Novartis, Pfizer and UCB Pharma. Chief Medical Officer for CARE Arthritis Limited. RL: grants from Abbott, Amgen, Centocor, Novartis, Pfizer, Roche, Schering-Plough, UCB Pharma and Wyeth. Consultancy fees from Abbott, Ablynx, Amgen, AstraZeneca, BMS, Centocor, GSK, Novartis, Merck, Pfizer, Roche, Schering-Plough, UCB Pharma and Wyeth. Honoraria/speakers’ bureau from Abbott, Amgen, BMS, Centocor, Merck, Pfizer, Roche, Schering-Plough, UCB Pharma and Wyeth. MR: consulting fees from UCB Pharma. Honoraria/speakers’ bureau AbbVie, Eli Lilly, Janssen, Novartis and UCB Pharma. Participation on a Data Safety Monitoring or Advisory Board for AbbVie, Eli Lilly, Janssen-Cilag, Novartis and UCB Pharma. LB, BH, TK, MK, SEA: employees and stockholders of UCB Pharma. AD: grants from Abbvie, Eli Lilly, GSK, Novartis, Pfizer and UCB Pharma. Consulting fees from AbbVie, Amgen, Aurinia, BMS, Boehringer Ingelheim, Celgene, Eli Lilly, GSK, Janssen, MoonLake, Novartis, Pfizer and UCB Pharma. Speaker for Janssen, Novartis and Pfizer.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.