Article Text

Abstract
Objective The lifetime recurrence rate (RR) of axial spondyloarthritis (axSpA) among first-degree relatives (FDR) and the effect of proband’s gender, HLA-B27 and radiographic status is unclear. Our 35-year-follow-up family study has enabled these issues to be addressed.
Methods In 1985, 363 ankylosing spondylitis (AS) probands (members of the Swiss AS Patient Society) and 806 FDR recruited into the study, completed questionnaires regarding axSpA manifestations, underwent a physical examination and most also underwent pelvic radiography and HLA-B27 typing. At follow-up in 2018–2019, of the former participants whose current addresses could be retrieved, 162 had died and 485 (125 patients with AS plus 360 FDR) completed a postal questionnaire.
Results At follow-up, 48 of 177 HLA-B27(+) FDR had developed axSpA, an RR of 27.1% (95% CI 20.6% to 33.7%). 27/148 (18.2%) children of AS probands (modified New York (mNY) criteria) were affected versus 2/50 (4.0%) children of non-radiographic axSpA probands (p=0.0138, OR=5.36; 95% CI 1.23 to 23.40). Children of female probands were more often affected (12/22; 54.5%) than of male probands (15/78; 19.2%) (p=0.0003; OR=4.89; 95% CI 1.96 to 12.23). This increased risk applies equally to sons and daughters.
Conclusion The lifetime RR of axSpA for HLA-B27(+) FDR is substantial (27.1%), and disease severity (as defined by radiographic sacroiliitis by the mNY criteria) is an additional risk factor. Affected mothers pass on the disease significantly more often to their offspring than do affected fathers. These findings may lead to better assessment of lifetime risk for axSpA in the offspring. Moreover, investigation of this gender effect may uncover additional putative disease susceptibility factors.
- Ankylosing Spondylitis
- Epidemiology
- Low Back Pain
- Polymorphism, Genetic
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Familial occurrence of axial spondyloarthritis (axSpA) is well known.
The effect of proband’s gender, HLA-B27 and axSpA radiographic status on disease recurrence among first-degree relatives (FDR) is unclear.
WHAT THIS STUDY ADDS
The lifetime recurrence rate (RR) for HLA-B27(+) FDR is substantial (27.1%).
Risk for offspring of HLA-B27(+) ankylosing spondylitis (AS) mothers is significantly higher compared with offspring of HLA-B27(+) AS fathers.
The recurrence risk of relatives of probands with AS (by modified New York criteria) is much higher than that of relatives of probands with non-radiographic axSpA.
Our findings suggest that female AS probands are genetically ‘enriched’ with disease susceptibility genes.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
The substantially high lifetime RR of axSpA for FDR of AS probands necessitates greater emphasis on patient education and counselling.
There is a greater need for earlier diagnosis among FDR for better outcome due to increasing availability of more effective drugs.
It is important to further investigate this gender effect for uncovering additional putative disease susceptibility factors and for better assessment of lifetime risk for axSpA.
Introduction
Ankylosing spondylitis (AS) occurs worldwide; its prevalence in northern Europe approaches 0.5%,1–8 and 85%–95% of these patients possess HLA-B27 versus approximately 8% of the general population.9–11 Twin studies have shown concordance rates of 25%–75% in monozygotic versus 4%–15% in dizygotic twins.12 13 The reported recurrence rate (RR) for the first-degree relatives (FDR) of patients with AS has ranged between 4% and 11%.14 We had observed a 21% RR for HLA-B27-positive (HLA-B27(+)) FDR of HLA-B27(+) AS probands versus 1.3% for HLA-B27(+) adults in the general population.8 HLA-B27 is the strongest risk factor and contributes to ~20% of the heritability of AS,15 and polygenic risk scores (PRS) have been developed incorporating additional known risk factor.16 Higher prevalence of disease among men than women is seen in clinical practice, and men and women may differ in the likelihood of passing on the disease to their children.17 The relationship of the disease RR to age of the FDR, or to the proband’s gender and disease severity is insufficiently known, and the published reports are conflicting.17 18 We address these issues in our nationwide Swiss AS family study with a 35-years follow-up.
Methods
We assessed the RR of AS among FDR by gender of the index case (proband) considering their HLA-B27 status and the proband’s disease classification (as reflected by the presence of radiographic sacroiliitis). RR implies the proportion of FDR who develop the disease (occurrence); it does not mean exacerbation after remission.
Initial phase of the study (1985–1986)
In 1985, all members of the Swiss AS Patient Society were invited to participate together with their spouses and FDR, irrespective of health status. The study was performed in centres spread all over Switzerland after ethical approval from the University Hospital of Bern and informed consents were obtained from the participants.
Altogether, 1178 persons consented to participate, completed questionnaires on disease manifestations and underwent physical examination of their axial and peripheral joints by a rheumatologist. Blood samples were drawn for HLA typing and peripheral blood nucleated cells (PBNC) were stored. Consenting non-pregnant participants aged ≥18 underwent pelvic radiography unless a recent radiograph was available. All 1081 pelvic radiographs of 360 probands and 713 FDR and 8 spouses were assessed two times, blind to participants’ clinical and HLA-B27 status, by each of four experienced readers, that is, eight (sometimes nine) blinded readings for each sacroiliac (SI) joint. This could be performed only once for 46% of the 360 radiographs of the probands and 3% of 713 radiographs of the FDR because these radiographs were only available on-site at the time of participant’s physical examination in the local hospital. Overall, 17.2% of 1081 radiographs were read once, 0.4% 2–4 times, 3.2% 5–7 times and 79.2% 8–9 times.
The sacroiliitis score ranged from 0 (normal) to 4 (ankylosis) for each SI-joint assessment by a reader as per the modified New York (mNY) scoring system,19 Scores for a single SI joint were added and divided by the number of assessments (range 1–9). Scores below bilateral grade 2.0 were considered not fulfilling the mNY criteria, as did unilateral below grade 3.0 sacroiliitis. Interobserver and intraobserver reliability were assessed by evaluating a subset of 243 pelvic films. Observers read films two times in sets of 40–50 radiographs. The interval between both readings was ≥7 days.20
In this paper, the term axial spondyloarthritis (axSpA) comprises the full spectrum of disease, that is, both AS by mNY and non-radiographic axSpA (nr-axSpA). In 1986, participants received their study results.
Follow-up study (2018–2019)
In January 2018, the ethics committee of the Swiss Kanton Bern approved the follow-up study (#2017–00536). The former participants were asked to complete a 157-item postal questionnaire on features of axSpA that particularly dealt with symptoms at lumbar and gluteal region, thoracic spine and anterior part of the chest. Questions also addressed symptoms suggesting episodes of acute anterior uveitis. Apart from current or past axSpA manifestations, it was questioned whether between 1986 and 2019, a diagnosis of AS or axSpA was established by a Swiss rheumatologist and, if so, whether diagnostic imaging has been performed. The most decisive step to perform the follow-up study was to find out whether the former participants were still alive and, if so, to trace their current postal addresses. Starting in April 2018, up to five mailings to many Swiss city or village administrations were sent to obtain information dealing with current address of the former study participants and any deaths (including year of death).
In the spring of 2019, altogether 791 letters providing detailed information about the follow-up study were sent to updated, supposedly correct, addresses. Participants who provided written informed consent were mailed the questionnaire, and a reminder was sent to those who did not return the questionnaire. The data from all 485 questionnaires (of consenting participants) that had been returned by December 2019 were coded and anonymously stored in an Excel database for further analysis.
Ascertainment of diagnosis
The diagnosis AS in the initial study (1985–1986) is based on both the clinical findings and the evaluation of the—blindly read—pelvic radiograph described above. Probands and FDR were considered to have AS if they met the mNY criteria. Those with a clinical diagnosis of axSpA but not meeting the mNY criteria were deemed to have nr-axSpA. For the follow-up (2018–2019) study, we labelled all new patients found during the follow-up study as having axSpA (they have either radiographic axSpA (AS) or nr-axSpA) because recent SI joint images of all those reportly diagnosed to have AS by their rheumatologists were not available for us to review.
Polygenic risk score
From the PBNC samples, stored since 1985, PRS were calculated for 124 male and 24 female AS (mNY) probands. Genotyping was performed using the Illumina Core-Exome single-nucleotide polymorphism (SNP) microarray, as previously reported.16 Briefly, the PRS is a numeric summary score that reflects an individual’s estimated genetic predisposition for a given trait and can be used as a predictor or diagnostic biomarker for the disease of interest (here axSpA).
Statistical analysis
Counts were compared by χ2 testing, and the results are expressed as p values. ORs were calculated together with 95% CI.
Patient and public involvement
Two patients/coauthors were fully involved in the study.
Results
Initial phase of the study (1985–1986)
Altogether 1178 persons (630 men (53.5%) and 548 women (46.5%) participated in the family study: 363 AS probands, 806 FDR and 9 spouses. Table 1 and online supplemental flowcharts 1 and 2 provide their demographic data, together with radiographic results and HLA-B27 status. Considering reading of the pelvic radiographs, the interobserver and intraobserver reliability coefficients were 0.865 and 0.903, respectively.
Supplemental material
Demographic data of probands with AS, their relatives and spouses by HLA-B27 status and presence of sacroiliitis by the modified New York criteria
Among the 1136 participants who could be tissue typed, 671 (59.1%) were HLA-B27(+), and among the 358 probands who could be tissue typed, 308 (86.0%) were HLA-B27(+). The mNY criteria for AS19 were fulfilled almost two times as often (81%) by HLA-B27(+) versus HLA-B27(−) probands (44%), indicating th
at the latter group had significantly more often nr-axSpA (p<0.00001) (table 1 and online supplemental flowchart 1).
Among FDR sacroiliitis by mNY criteria was only seen in HLA-B27(+) relatives of HLA-B27(+) probands because 14/308 (4.5%) HLA-B27(+) relatives had AS, versus none of the 278 HLA-B27(−) relatives (p=0.00032) and none of the 85 relatives of HLA-B27(−) probands (p=0.0453) (table 1, online supplemental flowchart 2).
Follow-up study (2018–2019)
The Swiss village administrations reported that among the 1178 participants of the 1985–1986 baseline study, 162 had already died (123 probands and 39 FDR), 531 former participants could not be traced or declined participation (online supplemental table 1). The remaining 485 persons (125 probands and 360 FDR) agreed to participate in the follow-up study and completed the postal questionnaire (table 2, online supplemental flowchart 3). The response rate on the invitational letter was 61.3% (485/791). Of the 1178 participants of the 1985 baseline study, 485 (41.6%) took also part in the 2018–2019 follow-up study, whereas ≥162 (13.8%) had already died.
Supplemental material
Demographic data of probands and relatives participating in the Swiss AS follow-up family study by HLA-B27 status and presence of sacroiliitis by the modified New York criteria at baseline (1985)
Recurrence rate
AxSpA was present in 45 FDR; (42 of them HLA-B27(+); 17/67 (25.4%) men and 25/95 (26.3%) women); 38 of them did not have sacroiliitis in 1985 (or they did not undergo pelvic radiography because they then were ≤18 years or pregnant). The remaining seven were already diagnosed in 1985 as having AS (mNY). In contrast, axSpA occurred in only 3/184 (1.6%) HLA-B27(−) FDR (table 2, online supplemental flowchart 3).
Table 3 shows the lifetime RR (with 95% CI) for the HLA-B27(+) FDR to develop axSpA. This risk increased from 4.5% in 1985 (mean age 26.64 years) to 24.6% 35 years later in 2019 (when their mean age was 60.39 years). The lifetime RR estimate for HLA-B27(+) FDR increases to 27.1% (95% CI 20.5% to 33.6%) (table 3) by including all patients with AS who, apart from the proband, had already been diagnosed to have AS at study entry in 1985.
Risk for HLA-B27 positive FDR to have AS by the modified New York criteria at baseline, or to have axSpA at follow-up
Considering children, 27/148 (18.2%) children of AS (mNY+) probands were affected versus 2/50 (4.0%) children of nr-axSpA probands (p=0.0138, OR=5.36; 95% CI 1.23 to 23.40).
Effect of gender
For siblings of probands, the likelihood of having the disease is not influenced by gender of the family proband because 12/90 (13.3%) siblings of male probands versus 8/46 (17.4%) siblings of female probands were affected (p=NS).
However, the risk of developing axSpA among the offspring of HLA-B27(+) AS (by mNY criteria) parents is gender dependent because women probands are more likely to have at least one child also affected with axSpA than their male counterparts (p=0.02, OR=3.15; 95% CI 1.13 to 8.85) (table 4 and figure 1). Children of these women probands are significantly more often affected with axSpA than children of comparable fathers (p=0.0003 OR=4.89; 95% CI 1.96 to 12.23) (table 5). This gender effect (mothers transfer a higher risk to their offspring than fathers) applies equally to both sons and daughters of HLA-B27(+) AS (mNY+) parents (table 6). In contrast, this risk for HLA-B27(−) FDR is very low because only 3/184 (1.6%) HLA-B27(−) FDR acquired axSpA among all HLA-B27(−) FDR (of both HLA-B27(+) and HLA-B27(−) AS probands). The first of these three cases is that of a women and the HLA status of the proband is unknown because he never participated in the study. The second case is the son of an HLA-B27(−) male proband; at baseline the father (and the son) had no sacroiliitis, and there is no family history of psoriasis and inflammatory bowel disease. The third case, most interestingly, is that of an HLA-B27(−) sister whose HLA-B27(+) brother has AS, and their HLA-B27(+) father is unaffected.

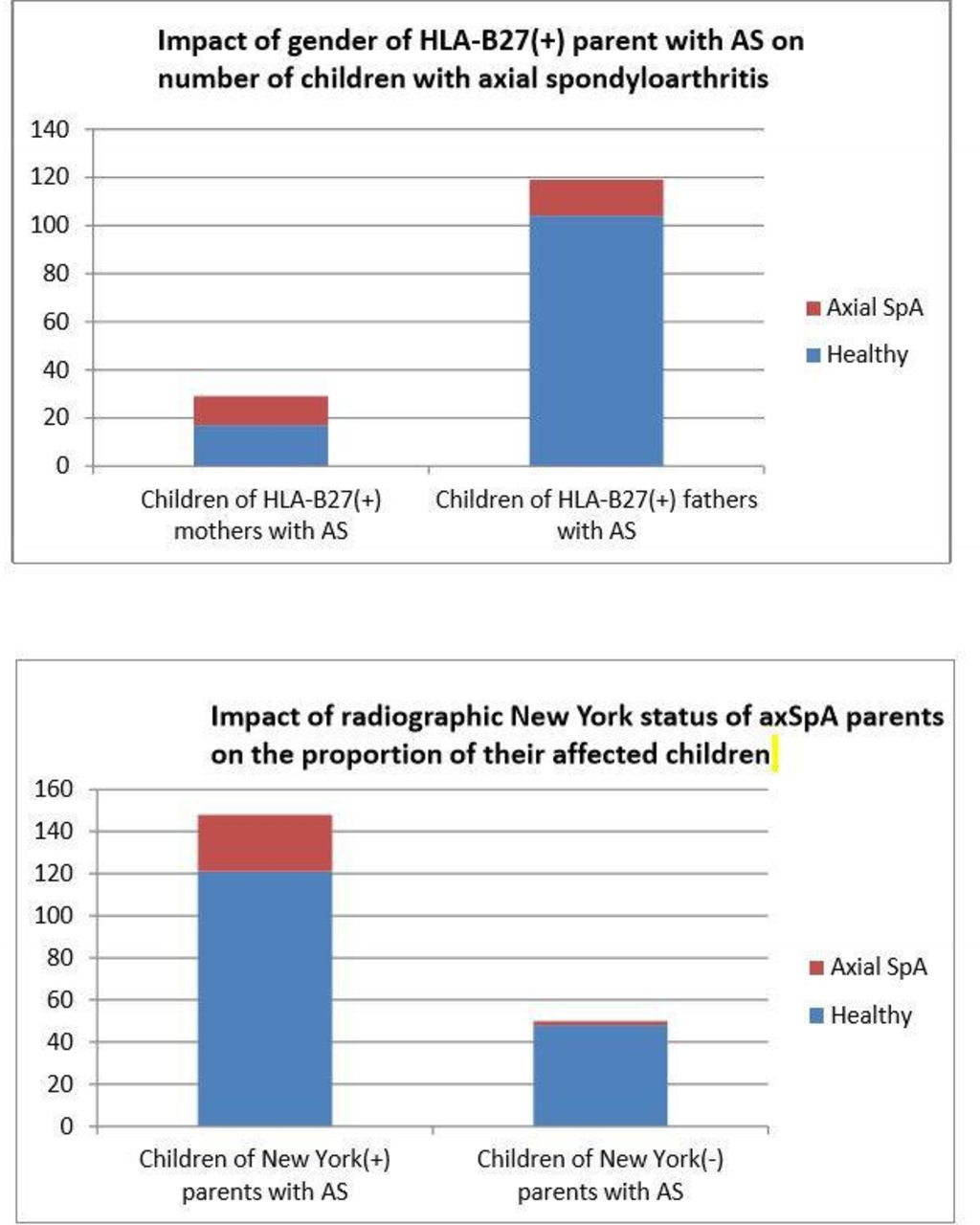{kind=link}
(A) A total of 185 (74.9%) male and 62 (25.1%) female HlA-B27 positive probands with ankylosing spondylitis (AS) meeting the radiographic New York criteria for sacroiliitis participated in the follow-up study. Mothers with AS had 29 children and fathers with AS had 119 children. The figure shows that 12 of 29 (41.4%) children of HLA-B27 positive AS mothers versus 15 of 119 (12.6%) children of HLA-B27 positive AS fathers have axial spondyloarthritis (axSpA) at follow-up (p=0.00032, OR=4.89 with 95% CI 1.96 to 12.23). At follow-up, all children were ≥45 years of age. (B) A total of 269 of 360 (74.7%) probands with AS met the radiographic New York criteria for sacroiliitis, whereas 91 (25.3%) had non-radiographic axial spondyloarthritis (nr-axSpA). The bar graphs show that 27 of 148 (18.2%) children of AS parents (meeting the radiographic New York criteria) versus 2 of 50 (4.0%) children of parents with nr-axSpA (p=0.0138, OR=5.36 with 95% CI 1.23 to 23.40) develop axial SpA. All children were ≥45 years of age at follow-up.
Mothers with HLA-B27 positive AS are more likely than HLA-B27 positive fathers with AS to get at least one child with axSpA*
Children of HLA-B27 positive mothers with AS are more likely to have axSpA than children of HLA-B27 positive fathers with AS
Proband’s New York radiographic and HLA-B27 status and proband’s gender strongly affect likelihood of axSpA among offspring
Effect of disease severity
The parent’s gender-specific risk for their offspring to get axSpA is most evident if the HLA-B27(+) AS proband meets the mNY criteria for the disease (table 6 and figure 1). Therefore, the parent’s disease severity (considering the presence of radiographic evidence of sacroiliitis to be an indicator of disease severity) enhances the risk associated with the inheritance of HLA-B27.
Gender and genetic risk score
To explore whether the observed higher parent-to-child disease recurrence for female than for male AS (mNY+) patients might be explained genetically, we compared mean PRS of 42 female HLA-B27(+) AS patients (0.418) with the mean value (0.376) of 124 male counterparts. The difference is not statistically significant.
Discussion
This Swiss AS family study spanning 35 years provides four important findings.
First, HLA-B27(+) AS probands were significantly more likely to have radiographic axSpA (ie, AS) than HLA-B27(−) probands. The mean ages of both groups are comparable (table 1). In our previous study, there were no differences in age at onset, functional class, degree of deformity, pain, severity of X‐ray changes or frequency of peripheral joint involvement between 63 HLA-B27(+) and 15 HLA-B27(−) AS patients,21 although subsequent studies have shown that HLA-B27(−) patients have later age of symptom onset and diagnosis.22 23 Therefore, taking the structural changes due to radiographic sacroiliitis as a measure of disease severity, one may extrapolate that on average HLA-B27(+) axSpA, patients have more damage than HLA-B27(−) patients in the context of familial occurrence of the disease. Consistent with this, a recent study reports that HLA-B27(+) FDR contrary to HLA-B27(−) FDR having early signs or symptoms progress to clinical disease.24 However, once patients develop radiographic AS, the extent of spinal damage has been shown not to be influenced by HLA-B27 status.21 25
Second, the lifetime RR of axSpA for HLA-B27(+) FDR was 4.5% in 1985 (mean age of FDR 26.64 years), and it increased to 27.1% 35 years later in 2019 (mean age 60.39 years). This figure applies for the full spectrum of axSpA (95% CI 20.5% to 33.6%) when we included all patients, apart from the proband, who had already been diagnosed to have AS by the time they entered the study in 1985 (table 3). This 27.1% RR of the disease can be regarded to be complete (lifetime) because it is extremely uncommon to have onset of AS after age 45. This RR estimate compares well with our smaller cross-sectional Dutch family study comprising 61 HLA-B27(+) and 40 HLA-B27(−) FDR of 20 HLA-B27(+) AS probands,8 in which study 5 of 24 (21%) HLA-B27(+) FDR aged 45 years or older had AS (by mNY criteria).
Third, the likelihood for brothers and sisters of HLA-B27(+) probands for having axSpA is not significantly influenced by the gender of the family proband. However, the RR for offspring is clearly gender dependent as we observed that children of HLA-B27(+) AS mothers significantly more often suffered from axSpA than children of HLA-B27 (+) fathers with AS. Please, note that risk estimates shown in table 6 apply to all children of these parents and increase about twofold for the HLA-B27(+) children. It would, therefore, be about as high as 80% for HLA-B27(+) children of female AS parents and about 25% for HLA-B27(+) children of male AS parents. Since in clinical practice, AS occurs more often in men, the genetic threshold for women to develop AS might be higher (they seem to be genetically ‘enriched’ with disease susceptibility genes) and that higher genetic load increases the recurrence of disease among both their sons and daughters. This is consistent with genetic models of polygenic diseases, where it is hypothesised that risk of the disease follows a normal distribution in the population, with those carrying more than a threshold of genetic risk ultimately developing the disease26 (further discussed below).
Finally, we observed that the RR of FDR of probands with AS is much higher than those with nr-axSpA (table 6). This is consistent with studies assessing the utility of PRS in AS compared with nr-axSpA, where the ability of the PRS to discriminate between clinically unaffected chronic back pain patients and AS patients was much higher than with nr-axSpA patients, consistent with nr-axSpA being less heritable and therefore less familial.27 Indeed, the RR of axSpA in children of HLA-B27(+) nr-axSpA probands (1/25=4%) is similar to the prevalence of AS among HLA-B27 carriers in the general community,8 28 29 suggesting that the nr-axSpA itself carries little additional familial risk other than HLA-B27 in those that carry this allele.
We are aware of some potential issues that may be raised about our follow-up analysis. One might question the potential for ascertainment bias. At baseline (1985), the diagnosis was based on full examination by a rheumatologist and blinded assessment of sacroiliitis according to the mNY criteria.19 Radiographs were read up to eight or nine times by up to four investigators who were blinded as to participant’s HLA-B27 status and clinical findings. This resulted in high specificity as none of the HLA-B27(−) FDR was found to have (false-positive) sacroiliitis. But in the follow-up study, the diagnosis, established by Swiss rheumatologists, was reported by the FDR, it was not possible to obtain recent radiographs or MRI of the SI joints of the ‘new’ cases, except for two of them. However, by using their responses to the questionnaire, 38/42 (90.5%) HLA-B27(+) FDR with axSpA met the ASAS classification criteria for axSpA, that is, they had either AS or nr-axSpA.30 31
The probands in our study were members of a patient society. They and their FDR volunteered to participate in the family study. A possible weakness of this kind of investigations is the potential of volunteer (selection) bias. People who have developed a certain disease might be more likely to participate in a study on their disease. FDR who had developed axSpA after the baseline study might be more inclined to participate in the follow-up study, leading to inflation of the true incidence of disease. On the other hand, studies based on registries—such as the one discussed later18—are not liable to volunteer bias, but suffer from other sources of confounding factors, such as misclassification and incomplete registration. These biases may arise, for example, when cases in a primary care setting are not referred to hospital or if there is a nationwide shortage of rheumatologists.
Our reported RR is unlikely to be a biased overestimation of the true risk. A bias would lead to a higher ratio of HLA-B27(+)/HLA-B27(−) FDR at the follow-up study when compared with the baseline study. The HLA-B27 status of the FDR was not known at the start of the study in 1985. However, the ratio between HLA-B27(+) FDR and HLA-B27(−) FDR of HLA-B27(+) AS probands has not changed; it was 1.17 in 1985 and 1.16 at follow-up (tables 1 and 2).
We report that HLA-B27(+) AS mothers (mNY+) are ~3 times more likely to pass on the disease to sons and daughters than HLA-B27(+) fathers, suggesting a higher genetic load among females AS probands (table 7). Our results are supported by a British family study of 4400 AS patients seen at an AS hospital and/or members of the British national patient society (National Ankylosing Spondylitis Society)17 and a Swedish nationwide registry-based family study.18 All show that risk for children to develop axSpA is greatest if the parent is woman. Furthermore, the British and Swedish studies also show that significantly more sons than daughters of AS fathers develop the disease. In Sweden, mothers as compared with fathers had more frequently an affected daughter.18 The British study also assessed the effect of disease onset below age 21 in female probands on their offspring; 5/16 (38%) children developed axSpA, in contrast to 3/39 (8%) children of fathers.17 These findings are in line with our results (41.4% for children of mothers vs 12.6% children of fathers). They also reported a higher rate of affected sibs if the proband is women; numbers in our study might have been too small to show such an effect. The prevalence of AS among the FDR in the Swedish study ranged between 2.1% and 3.6%, contrasting with a 4%–11% prevalence observed in European countries,14 and it was 0.07%–0.19% among Swedish controls (comparing well with the 0.1%–0.4% figure from the literature.1–8
Recurrence rate of axSpA among siblings and children in three studies
The disease RR for the offspring of HLA-B27(+) mNY +AS mothers is higher compared with male probands, but PRS of the female probands while 11% higher than the PRS of male probands is not statistically significantly increased. What about a possible explicatory role of the X-chromosome and (by definition maternal) mitochondrial DNA? Since in clinical practice, AS occurs more often in men, the genetic threshold for women to develop AS might be higher (they seem to be genetically ‘enriched’ with disease susceptibility genes), and that higher genetic load increases the RR among both their sons and daughters. This is consistent with genetic models of polygenic diseases, where it is hypothesised that risk of the disease follows a normal distribution in the population, with those carrying more than a threshold of genetic risk ultimately developing the disease.26 At this point, the actual genes involved in the difference in genetic risk between genders are not clear, as we have discussed elsewhere.32 Differences in recurrence of disease in offspring patients in highly heritable conditions like AS could potentially occur where there are significant X-chromosome or mitochondrial encoded susceptibility variants involved. The PRS employed here only uses autosomal SNPs and does not include X-chromosome markers. Substantial X-chromosome involvement would lead to female children of male AS patients being more likely to develop AS; evidence to date suggests that the converse is true.17 Linkage studies have excluded major genetic effects on the X-chromosome in AS,33 association studies of the X-chromosome are in progress. Mitochondrial genetic diseases are transmitted from affected mothers rather than affected fathers, consistent with our observation of increased risk in offspring of affected mothers. However, the gender ratio of affected children in offspring of mothers with mitochondrial diseases is typically equal, which is not consistent with the known gender bias in AS. Therefore, neither X-chromosome nor mitochondrial genetic variation are likely to be the dominant explanatory factor of the gender bias in AS in the general population along with the difference in RR of offspring of affected parents, whereas differences in the required genetic threshold to develop the disease can explain both these features of AS. Currently, one can only speculate about possible drivers of gender-related different thresholds, for example, epigenetic, hormonal or other environmental factors. Further studies comparing genetic associations in male and female AS patients will be required to investigate whether the threshold model is correct.
Finally, a family study with follow-up 35 years later is challenging. Some lessons learnt are provided in online supplemental table 2.
In summary, the lifetime RR of axSpA for HLA-B27(+) FDR is substantial: 27.1% (95% CI 20.6% to 33.7%). HLA-B27(+) female AS probands are significantly more likely to pass on the disease to sons and daughters than the HLA-B27(+) male AS probands. The risk increases twofold if the child inherits the HLA-B27 allele from the affected parent. Genetic susceptibility threshold for women to develop radiographic axSpA seems higher than for men, and it enables women to transmit the disease susceptibility to a higher proportion of their offspring. Independent of HLA-B27, offspring of patients with nr-axSpA is not at increased risk of axSpA. These findings are also relevant for patient education and counselling. It is important to further investigate this gender effect for uncovering putative additional disease susceptibility factors and for better assessment of lifetime risk for axSpA. Finally, to honour our teachers, long before the HLA-B27 era, studies had already reported an increased risk of disease for offspring of female AS patients.34 35
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Ethical Committee of Kanton of Bern, Switzerland#2017-00536. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We like to thank all patients, spouses and relatives for their kind cooperation and are grateful to the Swiss city and village administrations for retrieving current addresses. We also thank Hans-Ueli Rentschꝉ, MD, Hans Valkenburgꝉ, MD, Arnold Catsꝉ, MD, Herman Kroon, MD and Niklaus Gerber, MD for their contributions in performing the study. Caroline Kaegi provided helpful secretarial assistance. (ꝉDeceased)
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Deceased HB Deceased
Contributors All authors had full access to all of the data in the study and take responsibility for the data and the accuracy of the data analysis. Concept and design: SMvdL, MAK, HB, HvZ and MB conceived the study. SMvdL, HB and HvZ designed the questionnaire. Drafting of the manuscript: SMvdL, MAK and MAB. Genetical analysis: ZL and MB. Statistical analysis: SMvdL, ZL, MKK and MB. Obtained funding: SMvdL and MAB. Administrative, technical or material support: HvZ and PMV. Acquisition of data: SMvdL, HvZ and PMV. Critical revision of the manuscript for important intellectual content: all authors. Guarantor: SMvdL.
Funding The 1985 baseline study was funded by the Swiss National Fund, Schweizer Rück Insurance, and Ciba-Geigy, Switzerland. The 2018 follow-up study was funded/supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London and/or the NIHR Clinical Research Facility.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.