Article Text

Statistics from Altmetric.com
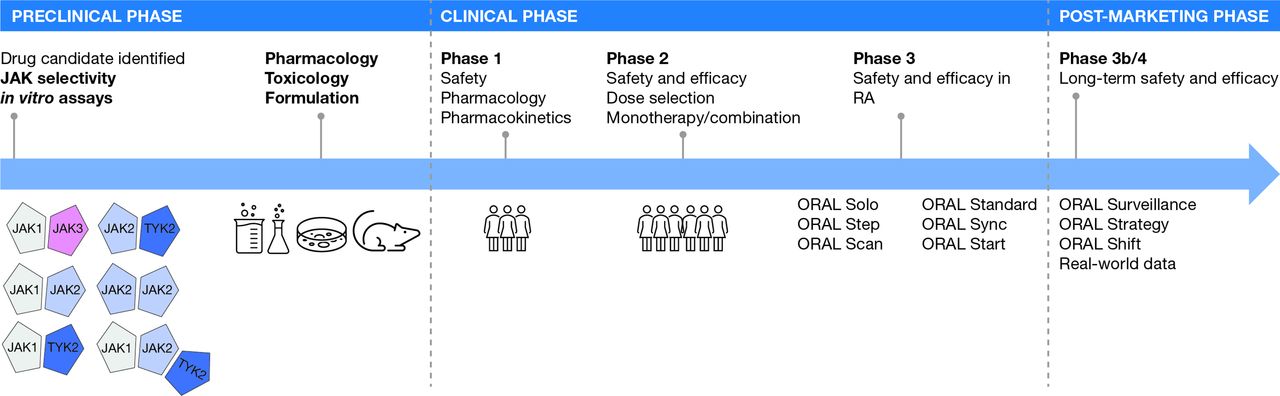
The discovery of Janus kinase (JAK) proteins in the early 1990s,1 which led to the approval of tofacitinib in rheumatoid arthritis (RA) decades later, serves as an illustrative case study of drug development (figure 1). The potential for therapeutic targeting of the JAK family first became apparent when it was found that patients with inactivating mutations in JAK3 developed severe combined immunodeficiency.2 Furthermore, heterozygous parents of these immunodeficient patients were immunocompetent,2 suggesting that the immune system could be modulated by partial inhibition of JAK3. Additional genetic mutations in JAK2 (activating) and tyrosine kinase 2 (TYK2; inactivating) were also found to be associated with immune phenotypes amenable to therapeutic blockade.3 4 However, quite how each of the four JAK family members (JAK1, JAK2, JAK3 and TYK2) contributes to health and disease was unclear in the 1990s, and while incredible progress has been made over the past two decades, many important questions about JAK biology remain unanswered.

{kind=link}
Development of tofacitinib, the first JAK inhibitor.In RA, the JAK pairing model here is a simplified representation and does not show cytokine or STAT pairing. The preclinical phase asked the questions: Are JAKs worth targeting for inflammation? Does the candidate bind and block all JAK isoforms, or is there selectively in silico? In vitro? In vivo? Can we optimise the candidate to improve effects? Are there off-target effects? What is the toxicology profile in relevant species? Does the candidate have ‘drug-like’ properties that would enable convenient dosing, preferably once-daily as a pill? The clinical phase defined the safety and efficacy of tofacitinib in patients with rheumatoid arthritis. JAK, Janus kinase; RA, rheumatoid arthritis; STAT, signal transducer and activator of transcription; TYK2, tyrosine kinase 2.
After the biological rationale for inhibiting JAK signalling became apparent, the next logical question was which JAK protein(s) to target. Early animal models suggested that deficiencies in JAK1 and JAK2 (and by extension, inhibition with medicines) would have more profound effects on the >50 JAK-dependent signalling pathways when compared with either JAK3 or TYK2 inhibition.5 6 These observations led to attempts for ‘selective’ inhibition of JAK isoforms, specifically aiming to spare JAK2 because of its role for growth factor signalling.6 However, questions continue to swirl about the clinical meaningfulness of ‘selectivity’ against a specific JAK isoform; in this short review, we attempt to address some of these issues.
The biochemical and cellular selectivity profile of a JAK inhibitor is defined in the preclinical phase
JAKs are enzymes, and JAK inhibitors generally work by competing with (adenosine triphosphate) ATP for the enzyme binding pocket.7 This ‘competitive binding’ strategy has enabled many drugs that target key enzymes to be designed and developed over recent decades, with the design process often guided by the three-dimensional structures of both the enzyme and the drug to find clever ways to fit a molecule into the binding pocket (akin to a key fitting into a lock). Importantly, however, for JAK inhibitors that bind to the active site, the ATP binding pockets of JAK1/2/3 and TYK2 are structurally very similar, so designing a molecule that targets one member of the JAK family without blocking the others (ie, true selectivity) has been quite challenging.6
Before any potential medicine is studied in humans, it must first undergo an extensive set of studies, often of increasing biological complexity. First, enzymatic assays test the ability of a drug to bind with and block the target; for all JAK inhibitors, this means carefully measuring the direct binding to each JAK member (usually done ‘in silico’ without any living cells). However, even these simple binding studies are critically dependent on the concentration of competing ATP in the experiment.7 The next step is to understand how these ‘biochemical’ effects in silico translate to living cells (that have JAK-dependent receptors); in vitro assessments are conducted under conditions that attempt to recreate physiological conditions—typically by exposing human blood leukocytes to relevant concentrations of the ‘selective’ JAK inhibitor. The assays used in characterising JAK selectivity have evolved over time, and today we have a better understanding of the molecular and cellular factors that may have impacted the results. In fact, initial descriptions of CP-690,550 (later called tofacitinib) mischaracterised the drug’s effects on JAK38 due to a limited appreciation of the assays used at the time to show selectivity.6 Multiple contemporary comparative in vitro and ex vivo assessments of approved JAK-inhibiting agents have been published9–12 and have generally reached similar conclusions: JAK inhibitors currently approved for use in rheumatology (tofacitinib,13 14 baricitinib,15 16 upadacitinib17 18 and filgotinib19) all potently inhibited JAK1, with some subtle differences around the effects on JAK210 or JAK3.9 Of interest, there are now JAK inhibitors in development that selectively avoid JAK1,20 21 minimising the effects of either immunosuppressive JAK-dependent pathways (eg, IL-6)20 or JAK2-dependent effects on growth factors.21 Some of the JAK inhibitors in development include deucravacitinib, an allosteric TYK2 inhibitor,21 22 and ritlecitinib, a covalent JAK3/TEC inhibitor.23 24 Targeted selectivity of JAK theoretically reduces side-effects by sparing certain JAKs (eg, JAK2); however, given that JAK inhibitors target the enzymatic active sites, off-target effects (eg, changes in haemoglobin levels), may be seen25–27 and presented in a dose-dependent manner.25 27
Ultimately, in the preclinical phase (in vitro and animal models), data are generated to support the potential efficacy of the drug for the treatment of a disease state and potential adverse events based on the on-target and off-target actions of the drug. Thus, the selectivity profile of a given JAK inhibitor, defined in the preclinical phase, can be used to generate hypotheses about the efficacy and the likely safety profile of the drug. However, this proposed profile remains hypothetical until the clinical phase of the drug development process begins.
The efficacy and safety profile of a drug is ultimately determined in human clinical trials
The first task when entering human studies is to test whether the drug is safe. Initial studies in a healthy population (phase 1) enable further investigation of the drug’s clinical efficacy in a focused patient population (phase 2). However, even after successful controlled clinical studies (phase 3) and approval for use as a medication, long-term safety and efficacy must be assessed in the real-world setting, via postauthorisation safety studies (phase 4) and analysis of real-world data (eg, registries or claims data).
Today, JAK inhibitors form part of the therapeutic armamentarium for several immune-inflammatory diseases, including RA, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, graft-versus-host disease, atopic dermatitis and ulcerative colitis.13–19 28–30 Treatment with these agents is efficacious, with rapid onset of action and the convenience of oral delivery or topical application.13–19 28–30 However, since approval of tofacitinib for RA in 2012,13 specific safety concerns associated with JAK inhibitors have emerged, namely reactivation of herpes zoster and serious infections.31–33 More recently, additional safety considerations include malignancies,34 venous thromboembolic events (VTE) and major adverse cardiovascular events (MACE).34 35
Data from ORAL Surveillance-like studies for other JAK inhibitors are needed
When tofacitinib was first approved for the treatment of RA, the US Food and Drug Administration (FDA) requested data from a dedicated, safety-focused controlled clinical trial to evaluate the long-term safety of the drug in patients with RA.36 How should such a study be designed to capture rare safety events or events with long latency? There are three factors to consider: (1) the study must include a sufficiently large number of patients; (2) it must run for a sufficiently long duration and (3) the chances of capturing events of interest can be increased by enriching the study population with risk for the events of interest. Accordingly, in the 2012 New Drug Application letter to Pfizer, the US FDA specifically stated that only a clinical trial (rather than a nonclinical or observational study) will be sufficient to assess signals of safety events, such as MACE, serious infections and malignancy’ and that the trial should be of sufficient size and duration to evaluate safety events of interest.36
ORAL Surveillance was a large, postapproval, open-label, safety event-driven clinical trial that was designed to compare the long-term safety of tofacitinib at two doses versus a tumour necrosis factor inhibitor (TNFi; adalimumab in North America, and etanercept in the rest of the world) in patients with moderate to severe RA despite methotrexate treatment.37 The study was specifically designed to assess the risk of MACE and malignancies (excluding non-melanoma skin cancer [NMSC]); therefore, in contrast to previous studies, the population was enriched to increase the likelihood of observing the required number of events within the projected 5-year study duration. Patients were required to be at least 50 years of age and have at least one additional cardiovascular (CV) risk factor to be eligible to participate.37 The study could only be declared completed when at least 1500 patients were followed for a minimum of 3 years each and a minimum of 103 adjudicated MACE and 138 adjudicated malignancies had been identified.37 Borne out of this specific design, ORAL Surveillance took 6 years to complete, included 4362 patients (of whom 1455, 1456 and 1451 received tofacitinib 5 mg two times per day, tofacitinib 10 mg two times per day and a TNFi, respectively), and showed that the incidences of co-primary endpoints MACE and malignancies, excluding NMSC, were higher with the combined tofacitinib doses (3.4% (98 patients) and 4.2% (122 patients), respectively), than with a TNFi (2.5% (37 patients) and 2.9% (42 patients), respectively).37 In February 2019, the tofacitinib dose of 10 mg two times per day was reduced to 5 mg two times per day after the DataSsafety Monitoring Board noted a higher frequency of pulmonary embolism among patients receiving tofacitinib 10 mg two times per day than among those receiving a TNFi.37 In addition, the board noted higher mortality with tofacitinib 10 mg two times per day with tofacitinib 5 mg two times per day or with a TNFi.37 The study findings have resulted in revisions to the product label,13 14 including the US Prescribing Information Boxed Warning, which has been updated to include MACE and revised for mortality, malignancies and thrombosis.13
Similar studies to ORAL Surveillance have not been completed for other JAK inhibitors, leading to an important data gap with respect to comparing the safety profiles of different JAK inhibitors. It should be acknowledged that post-marketing studies assessing the safety of baricitinib vs TNFi with respect to VTE in enriched RA patient populations are currently underway,28 29 and, in February 2022, the European Medicines Agency announced a safety review of JAK inhibitors in the treatment of chronic inflammatory disorders.38 The paucity of data is frustrating for both rheumatologists and patients alike, who are missing essential information on non-tofacitinib agents when trying to make an informed decision. One recent article from a group of clinicians even went so far as to compare the act of waiting for comprehensive safety data for all JAK inhibitors to Samuel Beckett’s play‚ Waiting for Godot!39
In the absence of dedicated studies, real-world data represent an important means of monitoring the safety profiles of JAK inhibitors and drawing comparisons with other treatment options. Data from the US CorEvitas (formerly Corrona) RA Registry showed similar incidence rates (IRs) of selected adverse events (including MACE, serious infection events, malignancies, VTE and death) for patients initiating tofacitinib and those initiating biological disease-modifying antirheumatic drugs (bDMARDs) over 5 years.40 IRs of herpes zoster were higher for tofacitinib initiators than for bDMARD initiators.40 In a separate study of US claims data, the risk of CV outcomes in real-world RA patients initiating treatment with tofacitinib was no higher than that in patients initiating TNFi (HR [95%] 1.01 [0.83 to 1.23]); although the same comparison in patients who were at least 50 years of age and had at least one additional CV risk factor (mimicking the ORAL Surveillance population) showed higher risk with tofacitinib treatment, though statistically non-significant (HR [95%] 1.24 [0.90 to 1.69]).41
So, when considering the risks vs benefits of JAK inhibitors in the clinic, does selectivity data from in vitro studies help us bridge the clinical data gap? It is possible for different JAK inhibitors to have unique efficacy and side-effect profiles. However, the missing clinical information cannot be extrapolated from preclinical in vitro or ex vivo studies; it has to come from ORAL Surveillance-like clinical trial data.
JAK signalling in patients is much more complicated than that suggested by preclinical selectivity data alone
Given that the biological system is much more complex than the assays performed in the laboratory, in vitro studies are only hypothesis-generating. In a biological setting, on ligand binding to the outside portion of transmembrane cytokine receptors, JAK proteins are brought together, cross-phosphorylate and activate each other.6 This pairing is an essential step in signal transduction by this protein family.6 The JAK/JAK pair forms a single unit and activates downstream transcription factors (ie, signal transducer and activator of transcription (STAT)).6 JAK1 is the most promiscuous of the JAK proteins and pairs with all other JAK family members, while JAK3 only pairs with JAK1. JAK2 is unique in its tendency to pair with itself (JAK2/JAK2).6 All molecules that inhibit JAK1 (including tofacitinib, baricitinib, upadacitinib, filgotinib and abrocitinib)13 15 17 19 28 impact the receptors signalling via JAK1/JAK2, JAK1/TYK2 and JAK1/JAK3 pairs, as well as JAK1/JAK2 or TYK2 (figure 1). Because JAK1 always pairs with another member of the family in carrying out its function, even a JAK1-selective agent will affect the signalling of cytokine receptors that are also dependent on JAK2, JAK3 or TYK2. To make things even more complex, there are >50 known JAK-dependent cytokine receptor signalling complexes with unique, but also overlapping, STAT-dependent signalling pathways, and there is the potential for additive or synergistic biological effects in cytokine regulation, including—importantly—immunosuppressive pathways.5 Although selective inhibition of JAK3 or TYK2, by sparing JAK1 and JAK2, will impact fewer cytokine pathways, it will nevertheless affect signalling networks shown to have important roles in immune diseases.5 To put it simply, JAK biology is complicated!
Making clinical predictions based on laboratory data is alluring. Understanding where in vitro selectivity studies fit in the drug development process, relative to clinical trials, can help clinicians contextualise safety and efficacy results. However, disentangling on-target effects that are unique JAK1 (or any JAK) is challenging given the signalling architecture (ie, heteropairing) and the vast complexity of the human immune system. Furthermore, while preclinical in vitro and animal studies, such as selectivity studies, help generate hypotheses about how a drug would behave in human patients, ultimately the efficacy and safety of a JAK inhibitor must be borne out of clinical trials, ideally head-to-head studies, supplemented by real-world evidence.
Ethics statements
Patient consent for publication
Acknowledgments
Editorial support, under the direction of the authors, was provided by Justine Juana, BHSc, CMC Connect, McCann Health Medical Communications and was funded by Pfizer, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461-4). The authors would like to thank Martin Dowty, Jean-Baptise Telliez, Tsung H Lin and Jose Rivas for their contribution in the development of this piece.
References
Footnotes
Contributors All authors contributed to interpretation of the literature and data discussed in this manuscript, contributed to the writing of the first draft of the manuscript and critically reviewed subsequent drafts of the manuscript. All authors read and approved the final draft for submission.
Funding This work was funded by Pfizer.
Competing interests MM and DAM are employees and shareholders of Pfizer. HS-K has received grants and/or research support from Novartis and Pfizer, and has acted as a consultant for AbbVie, Amgen, Biogen, Bristol-Myers Squibb, Celgene, Eli Lilly, Gilead Sciences, Hexal Sandoz, Hospira, Janssen-Cilag, MSD, Novartis, Pfizer, Roche and UCB.
Provenance and peer review Not commissioned; externally peer reviewed.