Article Text

Abstract
Macrophage activation syndrome (MAS) is a subtype of haemophagocytic lymphohistiocytosis (HLH), and a well-described complication of systemic juvenile idiopathic arthritis (SJIA), triggered by disease onset or flare, infection, or some medications. Here, we report a 20-year-old man with previously well-controlled SJIA, who developed first time MAS after acute Epstein-Barr virus (EBV) infection, with MAS recurrence due to a drug reaction, ‘3-week sulfasalazine syndrome’, secondary to prophylactic trimethoprim/sulfamethoxazole. Both episodes of MAS were minimally responsive to pulse corticosteroids. Initial EBV-driven MAS was treated with multiple doses of emapalumab prior to resolution, while MAS secondary to sulfasalazine-induced 3-week syndrome required the initiation of ruxolitinib. This case exhibits two rare but life-threatening causes of MAS/secondary HLH in a single patient and the difficulties in their diagnosis and management.
- Treatment
- Antirheumatic Agents
- Arthritis, Juvenile
- Biological Therapy
- Sulfasalazine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Episodes of macrophage activation syndrome (MAS) can have diverse triggers, including uncontrolled rheumatic disease activity, infections and medications.
WHAT THIS STUDY ADDS
Sulfasalazine-induced 3-week syndrome and DRESS are related syndromes that can develop secondary to sulfasalazine-related medications and induce MAS.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Sulfasalazine-containing medications are used for both rheumatological conditions and as infection prevention in immunosuppressed patients with rheumatic conditions. Sulfasalazine-induced 3-week syndrome is a rare and important entity to consider in the differential diagnosis for MAS, since indefinite discontinuation of the medication is an important part of treatment.
Introduction
Macrophage activation syndrome (MAS) is a frequent complication of rheumatic disease including systemic juvenile idiopathic arthritis (SJIA), both at disease onset or triggered later by flares or other disease processes, such as infection, malignancy, or medication reaction.1 Epstein-Barr virus (EBV) is the most common infectious trigger of MAS.2 Immunosuppression for both SJIA and MAS puts patients at risk for severe infection, and infectious prophylaxis is frequently used.3 We report a patient with long-standing SJIA who developed a first episode of MAS after acute EBV infection, as well as subsequent MAS recurrence due to ‘3-week sulfasalazine syndrome’, which occurred secondary to trimethoprim/sulfamethoxazole (TMP/SMX) prescribed as antimicrobial prophylaxis for the treatments used during his first MAS course.
Case report
We report a 20-year-old otherwise healthy Caucasian man, diagnosed with SJIA at the age of 15 years, after presenting with symptoms of quotidian fevers, evanescent rash, and polyarthritis. At that time he was started on canakinumab with subsequent resolution of symptoms without any complications. After 5 years of well-controlled disease, he developed 3 days of fever, joint pain, and petechial rash. The patient’s symptoms in combination with laboratory findings, including ferritin 6028 ng/mL, platelets 47×10∧9/L, fibrinogen 117 mg/dL, aspartate aminotransferase (AST) 142 unit/L, were consistent with MAS.4 He was admitted and treated with intravenous methylprednisolone 1000 mg daily for 5 days and clinically stabilised, but his neutropenia, lymphopenia, thrombocytopaenia and hypofibrinogenaemia did not improve (figure 1). His CXCL9 levels returned at 34 638 pg/mL (reference range ≤647 pg/mL) so emapalumab was initiated twice weekly with rapid improvements in his cytopenias and hypofibrinogenemia.5 6 Given that this was his first MAS episode after years of well-controlled disease, a thorough workup was performed for MAS triggers. Bone marrow biopsy was performed showing increased numbers of CD8 positive T cells, scattered haemophagocytosis and no overt evidence of leukaemia or lymphoma. EBV serology showed negative IgG and IgM but quantitative PCR returned significantly elevated at 501 400 IU/mL, consistent with a primary EBV infection as trigger for his MAS episode. With the initiation of emapalumab, the patient was started on acyclovir and TMP/SMX for antiviral and antimicrobial prophylaxis. His labs continued to improve (figure 1) and he was discharged on emapalumab two times weekly, continued canakinumab every 4 weeks and prednisone 60 mg two times per day.
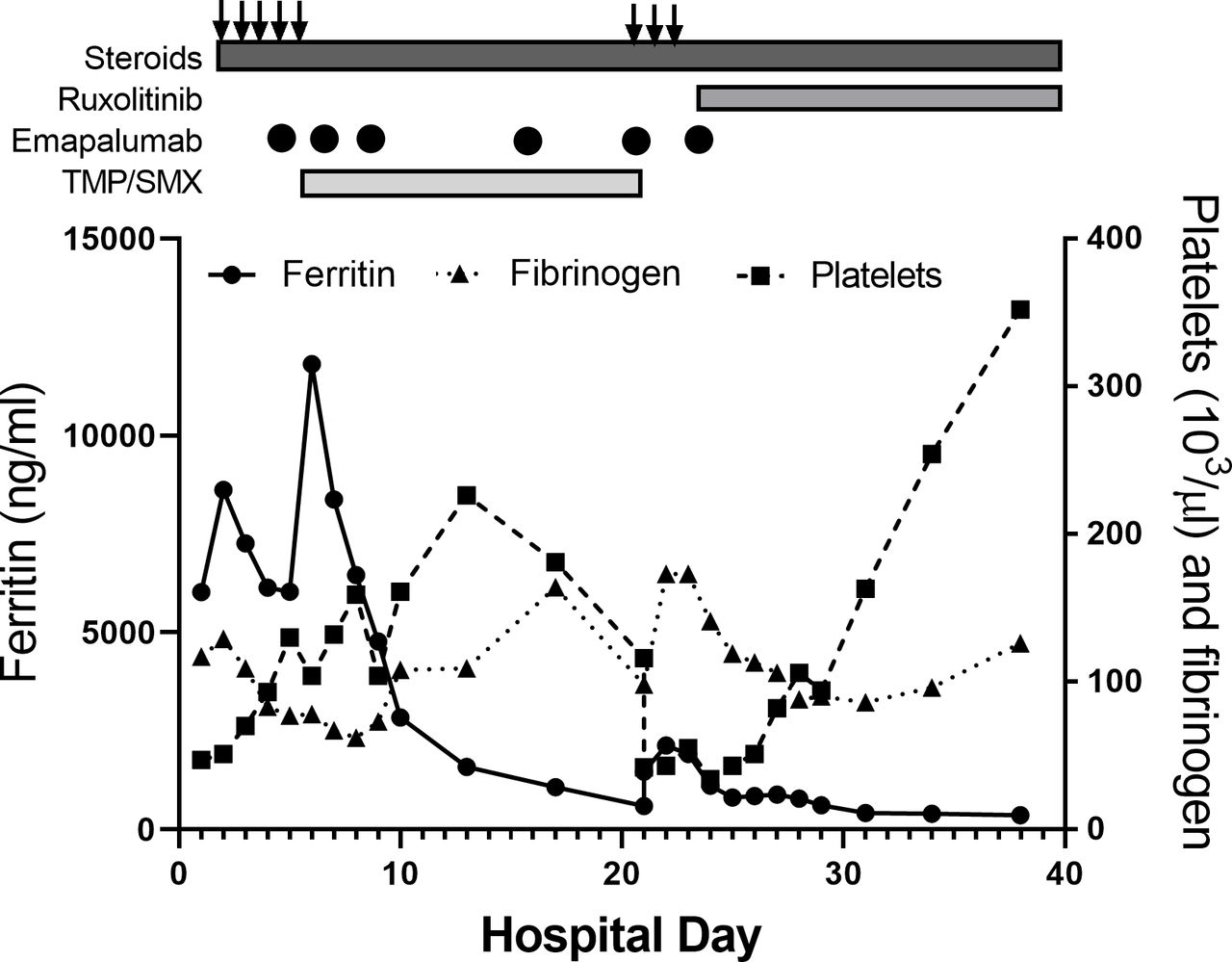
{kind=link}
Serial laboratory evaluation for patient from first episode of macrophage activation syndrome (MAS) through follow-up. Trend in ferritin, fibrinogen and platelet count in patient during and after hospitalisations for MAS. Treatments are shown above graph. Arrows represent intravenous methylprednisolone pulse therapy (1 g/day). Circles denote days of emapalumab infusion. TMP/SMX, trimethoprim/sulfamethoxazole.
Ten days after discharge and at the start of his third week of TMP/SMX, he developed fever and hypotension along with a diffuse erythematous rash on his neck, back, chest, arms and hands, as well as erythema of his conjunctiva with some facial swelling, while receiving emapalumab at our infusion centre. He was fluid resuscitated and further evaluated for causes of this acute decompensation. Comparison of labs before and after his TMP/SMX dose on the day of decompensation are shown in table 1 and demonstrate profound lymphopenia and thrombocytopaenia, worsening hyperferritinaemia and significantly elevated d-dimer. He was treated with empiric broad-spectrum intravenous antibiotics and simultaneously received intravenous methylprednisolone at 1000 mg daily × 3 days.
Serial laboratory evaluation for patient on day of acute decompensation, showing results before (7:00 hours) and after (19:00 hours) TMP/SMX administration
Differential diagnosis for his recurrent MAS included Steven-Johnson syndrome, sulfasalazine-induced 3-week syndrome, drug reaction with eosinophilia and systemic symptoms (DRESS), reactivation of EBV, or secondary infection. The timing and symptoms of his illness matched closely with that of both DRESS and sulfasalazine-induced 3-week syndrome, presenting approximately 3 weeks after starting TMP/SMX, a sulfasalazine-containing medication, and with symptoms of fever, rash, facial oedema, conjunctival oedema, haematological abnormalities and elevated transaminases.7–9 DRESS was considered given his symptoms; however, he only scored a 4 on the RegiSCAR scoring system for DRESS, and he did not fulfil Bocquet’s or the Japanese criteria.10 11 Bacterial and severe viral infections were ruled out with negative blood cultures and viral testing, and decreased EBV viral load. Unlike in many cases of sulfasalazine-induced 3-week syndrome and most DRESS, this patient did not manifest peripheral eosinophilia, but high-dose prednisone therapy may have prevented this typical response.
Treatment of sulfasalazine-induced 3-week syndrome and DRESS is discontinuation of the medication, so TMP/SMX was stopped, and prophylaxis was changed to intravenous pentamidine.7 As with his first episode of MAS, pulse steroids stabilised him clinically but did little to improve his lab abnormalities. Given his incomplete response to emapalumab, he was started on ruxolitinib, based on recent findings in a series of patients with EBV-driven MAS and secondary haemophagocytic lymphohistiocytosis (HLH).12 With this, he exhibited progressive improvement in his thrombocytopaenia and hypofibrinogenaemia (figure 1) and was discharged to complete a 28-day course of ruxolitinib and a tapering dose of prednisone. One month after his initial presentation of MAS, humoral response to EBV was checked with IgG titres and was positive. With known triggers for his episodes of MAS, genetic testing was not pursued.
Discussion
When faced with a patient with MAS, it is important to maintain a broad differential regarding possible triggers, as this will direct specific treatments. MAS triggered by new-onset SJIA or disease flare can often be effectively treated with high doses of anakinra, directed at the underlying autoinflammation.13 EBV-driven MAS/HLH is well documented and arises from a different mechanism than MAS triggered by SJIA. EBV typically infects and replicates within B cells; however, the virus can infect T and NK cells via CD21, leading to proliferation of cytotoxic CD8 T cells and cytokine storm with release of TNF and IFNɣ promoting widespread lymphohistiocytic activation.2 There are few reports on the use of emapalumab and ruxolitinib in the treatment of MAS/secondary HLH. Emapalumab, a fully human anti-IFN gamma monoclonal antibody, has recently been shown to be effective for HLH,5 and a recent case report exists where a patient with refractory EBV-associated secondary HLH was successfully treated with emapalumab.6
Sulfasalazine-induced 3-week syndrome and DRESS are similar and have overlapping features, but whether they are truly synonymous is unclear. Sulfasalazine-induced 3-week syndrome is a hypersensitivity reaction resembling DRESS that occurs secondary to exposure to sulfonamides which, in turn, causes MAS/secondary HLH. While well documented in patients with inflammatory bowel disease and in patients with rheumatoid arthritis being treated with sulfasalazine, this is the first reported case in a rheumatic patient receiving prophylactic TMP/SMX.14 15 Typical symptoms are fever, lymphadenopathy, dermatitis, haematological abnormalities and hepatitis around the third week of TMP/SMX initiation,7 and treatment requires the withdrawal of the offending agent and may require the use of other immune modulators.
DRESS is characterised by fever, skin eruption, internal organ dysfunction and haematologic abnormalities that develop after a new medication is introduced 1–8 weeks prior to the development of symptoms. Diagnostic criteria for DRESS includes the RegiSCAR, Bocquet’s and Japanese criteria.10 While there is overlap between the two syndromes, patients with sulfasalazine-induced 3-week syndrome may not meet criteria for DRESS, as seen in this patient. In addition, patients with DRESS tend to have a prolonged course even after withdrawal of medication, which this patient did not experience.10 Both syndromes are severe drugs reactions that require timely treatment and withdrawal of the offending agent.
Though MAS is a common complication of SJIA, this case highlights the importance of maintaining a broad differential for MAS triggers, even in patients with known SJIA. Also, while recognising the importance of antimicrobial prophylaxis in patients treated with multiple immunosuppressive medications, this case impresses that physicians and rheumatologists in particular, must be cognizant when using prophylactic TMP/SMX for their patients.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by CCHMC IRB 2018-2408. Participants gave informed consent to participate in the study before taking part.
Footnotes
Contributors MM and GSS are the primary pediatric rheumatology fellow and attending for the patient featured in this case report. MM wrote this case report with the guidance of GSS.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests GSS has received consulting fees from SOBI and Novartis.
Provenance and peer review Not commissioned; externally peer reviewed.