Article Text

Abstract
Objectives To report the impact of continued burosumab treatment on clinical laboratory tests of efficacy, patient-reported outcomes (PROs) and ambulatory function in adults with X-linked hypophosphataemia who continued from a 96-week phase 3 study into a 48-week open-label extension.
Methods Eligible participants from the phase 3 study continued on the burosumab regimen received at the end of the phase 3 study for a further 48 weeks (n=31). Some (not all) received compassionate burosumab treatment between the two studies (a period of 6–18 months). The primary efficacy outcome was fasting serum phosphate concentration; secondary outcomes were serum 1,25 dihydroxyvitamin D concentration, renal phosphate reabsorption, PROs and ambulatory function.
Results Improvements in fasting serum phosphate, serum 1,25 dihydroxyvitamin D and renal phosphate reabsorption at 96 weeks were maintained through the 48-week extension. Improvements were also maintained in stiffness and physical function measured using the Western Ontario and McMaster Universities Osteoarthritis Index, pain and fatigue endpoints measuring using the Brief Pain Inventory short-form and Brief Pain Inventory, respectively, and in ambulatory function (6-Minute Walk Test).
A post-hoc exploratory analysis exploring outcomes in participants who discontinued burosumab treatment between the studies (n=7) and those who received at least one dose (n=23) indicated that the benefits of burosumab on clinical laboratory tests of efficacy, PROs and ambulatory function may be lost when treatment is interrupted but recover over time when treatment is reinstated.
Conclusion Continued treatment with burosumab appears necessary for sustained clinical benefit.
Trial registration numbers Phase 3: NCT02526160; open-label extension: NCT03920072.
- Patient Reported Outcome Measures
- Therapeutics
- Outcome Assessment, Health Care
Data availability statement
Data are available upon reasonable request. Data are available on reasonable request from the corresponding author. All data relevant to the study are included in the article or as online supplemental file 1. The study protocol and statistical analysis plan for this study will be available on the relevant clinical trial registry websites with the tabulated results.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
In a Phase 3 trial adults with X-linked hypophosphataemia (XLH), burosumab treatment significantly increased serum phosphate concentrations compared with placebo over 24 weeks, with a sustained effect on phosphate homoeostasis. Patient-reported outcomes (PROs) improved significantly from baseline to 48 weeks, and clinically meaningful improvements in symptoms (pain, stiffness and fatigue) were sustained to 96 weeks
WHAT THIS STUDY ADDS
We assess the effect of a further 48 weeks’ burosumab treatment on clinical laboratory tests of efficacy, patient-reported pain, stiffness, physical function and fatigue and ambulatory function.
The correction of serum phosphate concentrations to above the lower limit of normal observed in the phase 3 trial is maintained with long-term open-label burosumab treatment.
Improvements in patient-reported pain, stiffness, physical function and fatigue and ambulatory function were maintained with long-term burosumab treatment, with clinically meaningful improvements observed across most PROs.
The benefits of burosumab appear to be lost if treatment is interrupted but return when treatment is reinstated.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The sustained positive outcomes support a role for long-term use of burosumab in improving physical function and quality of life for adults with XLH. Continued, uninterrupted burosumab appears to be necessary to sustain the clinical and biological benefits of treatment.
Introduction
X-linked hypophosphataemia (XLH) is a rare, inherited, lifelong disease in which excess fibroblast growth factor (FGF23) reduces the gastrointestinal absorption and renal reabsorption of phosphate indirectly by impairing the synthesis of 1,25-dihydroxyvitamin D3.1 The resultant chronic phosphate wasting has deleterious effects on bone growth and quality and causes deficits in muscle structure and function,2–4 which results in progressive accumulation of musculoskeletal disorders. The major feature of XLH in children is rickets, which causes bone deformities and short stature, resulting in pain and impaired physical functioning5; these skeletal abnormalities are locked in at the end of growth. Continued osteomalacia and misalignment of joints in adulthood can cause fractures, pseudofractures, early-onset osteoarthritis and enthesopathies, resulting in chronic musculoskeletal pain and stiffness, impaired physical functioning and mobility and fatigue.6–11
Until recently, the only treatment for XLH has been supplementation with oral phosphate and active vitamin D analogues; however, patients have to take multiple daily doses and undergo frequent monitoring and dose titration.12 In addition, patients experience gastrointestinal upset12 and are at risk of nephrocalcinosis, impaired renal function5 and tertiary hyperparathyroidism.13
Burosumab is a fully human monoclonal antibody against FGF23. Inhibition of FGF23 by burosumab results in increased absorption and reabsorption of phosphate, restoring serum phosphate concentrations to within the normal range.14 The efficacy and safety of burosumab in adults have been demonstrated versus placebo in the pivotal phase 3 study : serum phosphate concentrations increased to above the lower limit of normal (LLN), with concomitant improvements after 24 weeks’ treatment in stiffness and physical function, measured using the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC), and worst pain, measured using the Brief Pain Inventory short-form (BPI-SF).14 Follow-on treatment with burosumab for all patients showed continued benefits at 48 and 96 weeks.15 16
The further long-term efficacy of burosumab has now been evaluated in 31 European participants who, having received burosumab for 96 weeks in the phase 3 study, were invited to enrol into an open-label extension study so that they could continue treatment until 2021 or burosumab became commercially available. Some participants received compassionate burosumab treatment during the period between the two studies whereas others did not. The effect of the associated interruption to treatment in this subgroup of patients is also explored.
The primary objective of the current study was to assess the effect of continued burosumab treatment on the maintenance of serum phosphate concentration within the normal range. Secondary objectives were to assess the effects of continued burosumab treatment on additional clinical laboratory tests of efficacy, patient-reported outcomes (PROs) and ambulatory function.
An exploratory analysis was also conducted to compare the efficacy of burosumab in participants who did and did not receive compassionate burosumab treatment in the period between the two studies.
Participants and methods
Study design
The phase 3 randomised, double-blind, placebo-controlled efficacy and safety study was conducted at 25 centres in the USA, France, UK, Ireland, Italy, Japan and South Korea.5 14 16 Eligible participants were randomised 1:1 to receive burosumab or placebo, administered subcutaneously every 4 weeks for 24 weeks (online supplemental figure 1). Thereafter, all participants entered a treatment continuation period during which they received open-label burosumab. The final study visit was at week 96 at European centres (participants in the USA continued for up to 53 weeks further; participants in Japan and South Korea transferred to a long-term extension study (NCT04308096)).
Supplemental material
European participants from the phase 3 study were eligible to enrol onto an open-label extension (online supplemental figure 1); 34 participants from the phase 3 study from nine study centres met the eligibility criteria and were enrolled but 2 participants subsequently withdrew (online supplemental figure 2). At the data cut in January 2021, 31 participants had received up to 48 weeks’ further burosumab treatment in the open-label extension study. The current paper reports the results for these 31 participants from the phase 3 study baseline to week 96a and through the open-label extension study to week 48b. (The a and b suffixes are used to designate time points in the phase 3 study and open-label extension, respectively.)
Participants
Full inclusion and exclusion criteria for the phase 3 study have been reported previously.14 In summary, participants had to be adults (18–65 years old) with a confirmed diagnosis of XLH, biochemical findings consistent with XLH and a BPI-SF worst pain score ≥4. Major exclusion criteria included a recent history of traumatic fracture or orthopaedic surgery. Any therapies affecting phosphate metabolism had to be stopped at least 2 weeks before enrolment.
Investigators obtained written informed consent from each study participant.
Treatments
In the phase 3 study, participants received either burosumab (1 mg/kg) or placebo, administered subcutaneously every 4 weeks for 24 weeks; thereafter, all participants received burosumab through to week 96. Doses could be increased to a maximum of 1.8 mg/kg and were rounded to the nearest 10 mg (maximum 90 mg). Participants in the open-label extension study continued on the burosumab regimen they received at the end of the phase 3 study. However, there was an interval between the two studies when some, but not all, patients received compassionate burosumab treatment (mean 9 months; range 6–16 months). This reflects differences between countries in the regulations relating to the provision of compassionate treatment outside of clinical trials. The time to implement compassionate use in each country also varied. Treatment during the interim period was not captured.
Outcomes
Clinical laboratory tests, PROs, functional tests of efficacy and pain medication use reported by participants were assessed at the time points indicated in online supplemental figure 1. The primary efficacy outcome was the fasting serum phosphate concentration at the end of each dose cycle. Secondary outcomes were serum 1,25 dihydroxyvitamin D (1,25(OH)2D), and renal phosphate reabsorption (ratio of tubular maximum reabsorption rate to glomerular filtration rate (TmP/GFR)), calculated from fasting blood and urine measurements
PRO efficacy assessments (secondary outcomes) were measured using the WOMAC Index,17 BPI-SF18 and Brief Fatigue Inventory (BFI).19 The WOMAC Index includes scores for stiffness, physical function and pain, with a reference period of the past 48 hours. These scores can also be combined to a total score of 0–100, with higher scores indicating worse health. XLH-specific clinically meaningful changes have been defined.20 The BPI-SF, with a reference period of the past 24 hours, was used to generate scores for average and greatest Worst Pain (ie, average and greatest score over 8 days), Pain Severity and Pain Interference. Scores range from 0 to 10, with higher scores indicating worst pain severity or interference. XLH-specific clinically meaningful changes in scores have been defined.21 The BFI, with a reference period of the past 24 hours, was used to generate scores for average and greatest Worst Fatigue (average and worst fatigue over 8 days), Fatigue Severity, Fatigue Interference and Global Fatigue. Scores range from 0 to 10, with higher scores indicating worse fatigue severity or interference. XLH-specific clinically meaningful changes have been defined, except for BFI Fatigue Severity.22
Ambulatory function was assessed using the 6-Minute Walk Test (6MWT). Participants were instructed to walk the length of a premeasured course for six consecutive minutes and the distance covered measured in metres, as per published principles.23 Use of assistive devices was noted. Because of restrictions implemented during the coronavirus-19 (COVID-19) pandemic, some participants were unable to attend study centres at weeks 36b and 48b and therefore did not complete the 6MWT. The outcome assessments across the phase 3 and open-label studies are shown in online supplemental table 1.
Exploratory analysis of compassionate burosumab treatment between the studies
Following the review of the results for all participants, it was noted that not all participants had received compassionate burosumab treatment during the period between the two studies, even though compassionate treatment with burosumab was offered to bridge the gap between last dose in phase 3 study (week 96a) and first dose in the open-label extension (week 0b); the time between trials was 6–16 months. Post-hoc analyses were therefore conducted to evaluate the impact of treatment interruption on clinical laboratory values and PROs. One participant who did not complete the phase 3 study through to week 96a was excluded from this exploratory analysis because their time between trials without study drug was 28 months, which, given the small sample for this analysis, would likely skew the results in favour of burosumab. The remaining 30 participants were categorised into one of two groups: participants who received at least one dose of compassionate burosumab treatment in the period between the two studies, and those who did not receive any compassionate burosumab treatment in this period (figure 1).

Administration of burosumab during the interval between the end of the phase 3 study and the start of the open-label extension study. n=30; one participant who did not complete the phase 3 study through to week 96a was excluded from this exploratory analysis.
Statistical analysis
Statistical analyses were conducted in SAS .9.4 (SAS Institute, Cary, North Carolina, USA). The population for the primary analysis comprised all participants who enrolled in the phase 3 and open-label extension studies and who recorded at least one measurement after phase 3 baseline in the latter.
Continuous data are summarised as mean and SD, and categorical data as n (%). Clinical tests of laboratory efficacy are evaluated at each analytical time point alongside the LLN. PROs and functional endpoints are evaluated as change from phase 3 baseline to each analytical time point through to week 96 in the phase 3 study and in the open-label extension through to week 48. To maintain consistency with the analysis for the phase 3 study previously reported, a generalised estimating equation repeated-measures analysis was also performed. The model included treatment, actual randomisation stratification factor based on BPI Average Pain (except the model for BPI Worst Pain), region, visit and interaction of treatment-by-visit as fixed factors, adjusted for phase 3 baseline measurements. Compound symmetry was used as the covariance structure for the model, which specified constant variances for the assessments and constant covariances between the assessments over time. There were no statistical adjustments for multiplicity.
For the exploratory analysis on the impact of interruption to burosumab treatment on clinical laboratory tests of efficacy, Fisher’s exact test was used to compare the numbers of participants in the two groups with values above the LLN at the start of the open-label extension study. To assess the impact of burosumab treatment on PROs and ambulatory function, changes in PRO score/6MWT distance from phase 3 baseline to the start of the open-label extension study in the two groups were compared using the Mann-Whitney U test.
Results
Participants and treatment
The 31 participants included in this analysis had a mean age of 40.1 years (range 18.5–59.9) at baseline in the phase 3 study and 68% were women (table 1). The mean sex-specific percentile for height was 10.3%, indicating short stature due to impaired growth during childhood. All but 1 patient carried a heterozygous germline PHEX variant: 27 had pathogenic variants, 1 had likely pathogenic variants and 2 had variants of uncertain significance. One participant had no identified PHEX variation. At baseline in the phase 3 study, 65% of the participants had undergone orthopaedic surgery and 65% had a reported history of osteoarthritis. All participants had radiographic evidence of enthesopathy. More than half of the participants (55%) had evidence of nephrocalcinosis at phase 3 baseline. Most participants who took part in the open-label extension were using analgesics at phase 3 baseline (81%); 26% were using opioid analgesics. There were no significant differences between the phase 3 study population and the open-label extension study population in age, sex or BPI-SF worst pain at phase 3 baseline.
Demographics and characteristics at baseline in the phase 3 study for the whole study population and the subset who continued into the open-label extension
At the start of the open-label extension study (week 0b), burosumab doses ranged from 0.9 to 1.5 mg/kg (mean (SD) 1.0 (0.11) mg/kg; online supplemental table 2). The dose remained constant throughout the open-label extension for 20 participants. Seven participants had minor dose adjustments of ≤0.1 mg/kg and four had dose adjustments >0.1 mg/kg (0.2–0.5 mg/kg). Doses through the study are shown in online supplemental table 2.
No grade 4 or 5 treatment-emergent adverse events (TEAE) were reported and no participants experienced treatment-emergent serious adverse events (SAEs).
Clinical laboratory tests of efficacy
Over the phase 3 study and open-label extension, burosumab treatment was associated with initial improvements and then maintenance of serum phosphate and 1,25(OH)2D concentrations and phosphate reabsorption (TmP/GFR). At phase 3 baseline, the mean (SD) fasting serum phosphate concentration was below the LLN (0.6±0.02 mmol/L). Improvement was seen from week 12a (figure 2A) and the LLN was exceeded at week 36a (0.9±0.03 mmol/L). Correction of serum phosphate above LLN was maintained with continued burosumab treatment throughout the open-label extension study to week 48b (figure 2A). At phase 3 baseline, mean TmP/GFR was below the LLN (0.6±0.15 mmol/L). Improvement was seen at week 12a (0.7±0.26 mmol/L) and sustained through both studies, but the LLN was not reached at any time point (figure 2B). Mean serum 1,25(OH)2D was above the LLN at phase 3 baseline (80.0±29.8 pmol/L), increased at week 48a (99.4±30.4 pmol/L) and remained above the LLN throughout the phase 3 and open-label extension studies (figure 2C).
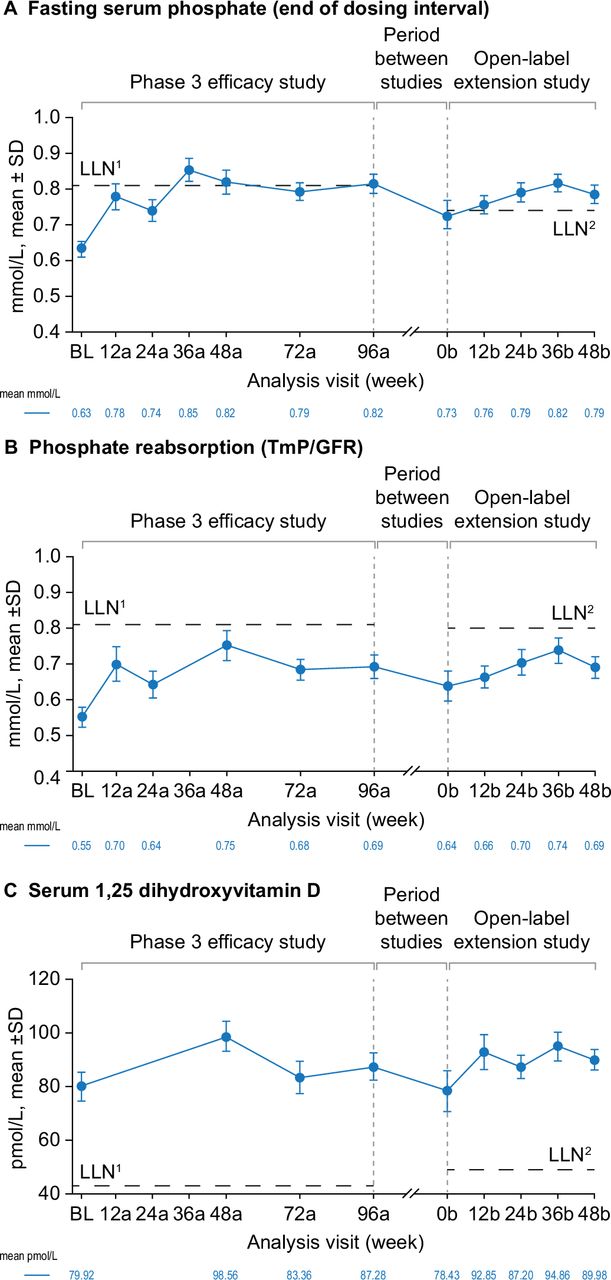
Effect of burosumab maintenance on clinical laboratory tests of efficacy. Data available for 31 participants at BL, 24 at week 48b for fasting serum phosphate and serum 1,25 dihydroxyvitamin D and 23 at week 48b for phosphate reabsorption (TmP/GFR). Analysis weeks in the phase 3 study and open-label extension are indicated by ‘a’ and ‘b’ suffixes, respectively. Samples from the two studies were measured at different central laboratories, with different LLN values for trough fasting serum phosphate (0.81 mmol/L in the phase 3 study (LLN1) and 0.74 mmol/L in the open-label extension (LLN2)); 1,25 dihydroxyvitamin D (43 and 48 pmol/L) and phosphate reabsorption (Tmp/GFR; 0.81 and 0.80–1.00 mmol/L). Serum 1,25 dihydroxyvitamin D and TmP/GFR were not measured at all analysis visits. BL, baseline; LLN, lower limit of normal range; TmP/GFR, ratio of tubular maximum reabsorption rate to glomerular filtration rate.
PROs
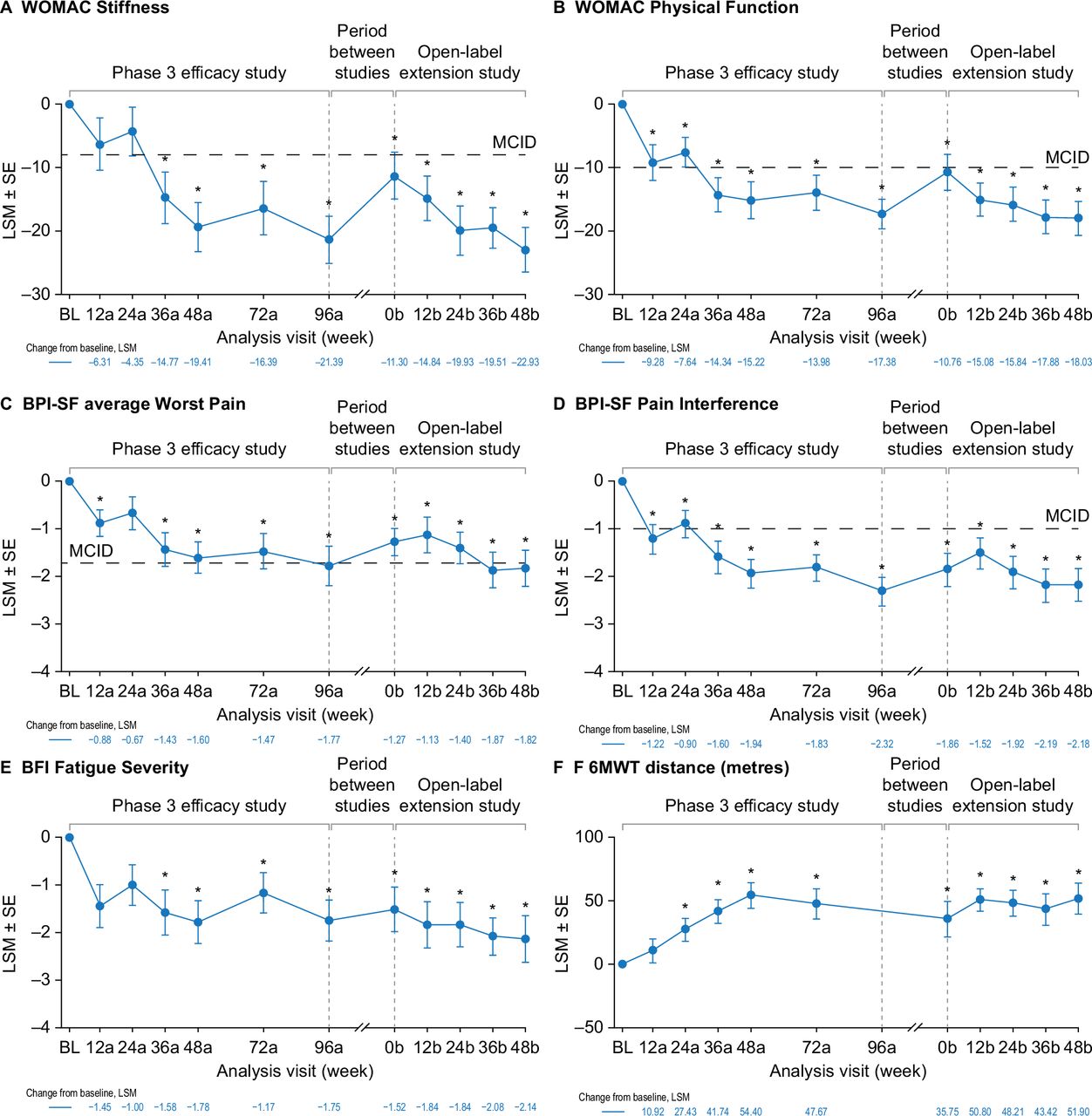
Burosumab was associated with sustained improvements in PROs over the phase 3 and open-label extension studies—WOMAC Stiffness (figure 3A), WOMAC Physical Function (figure 3B), BPI-SF average Worst Pain (figure 3C), BPI-SF Pain Interference (figure 3D) and BFI Fatigue Severity (figure 3E). WOMAC Stiffness scores had decreased significantly from phase 3 baseline at week 36a (least squares mean±SE, −14.8±4.04; p≤0.001) and at all subsequent time points in the two studies. Clinically meaningful improvements from baseline (≥8 point decrease21) were seen from phase 3 week 36a through to the end of the open-label extension study (figure 3A). WOMAC Physical Function scores had decreased significantly from baseline (improved function) at phase 3 week 12a (−9.3±2.79, p≤0.001) and at all subsequent time points through the two studies. Clinically meaningful improvements from baseline (≥10 point decrease21) were seen at phase 3 week 12a and from week 36a through to week 48b in the open-label extension study (figure 3B). BPI-SF average Worst Pain scores were decreased from baseline at phase 3 week 12a (−0.9±0.28; p=0.002) and at all subsequent time points in the two studies except for phase 3 week 24a. Clinically meaningful change from baseline (≥1.72 point decrease21) was seen at week 96a of the phase 3 study and weeks 36b and 48b of the open-label extension (figure 3C). BPI-SF Pain Interference scores had also decreased from baseline at phase 3 week 12a (−1.2±0.31, p≤0.001) and at all subsequent time points through the two studies. Clinically meaningful improvement from baseline (≥1.00 point decrease21) was seen at all study time points except phase 3 week 24a (figure 3D). The number of patients using pain medication decreased from 25 at phase 3 baseline (81%) to 15 (48%) at the end of the open-label extension. Opioid use decreased from 8 (26%) to 4 participants (13%) over the same period (online supplemental table 3). BFI Fatigue Severity scores had decreased from baseline at phase 3 week 12a (−1.5±0.45; p=0.001) and at all time points through to the end of the open-label extension study (figure 3E). Similar results were observed for other WOMAC, BPI-SF and BFI endpoints (online supplemental figure 3).

Effect of burosumab maintenance on PROs and 6MWT. Data available for 31 participants at BL, 28 at week 48b for WOMAC, 27 at week 48b for BPI-SF and BFI and 16 at week 48b for 6MWT. Analysis weeks in the phase 3 study and open-label extension are indicated by ‘a’ and ‘b’ suffixes, respectively. A decrease in score indicates improvement on the WOMAC, BPI-SF and BFI. An increase in distance indicates improvement on the 6MWT. BPI-SF and BFI data were captured at a single site visit and were not completed as part of a patient diary at weeks 72a and 96a. *p<0.05 for LSM change from BL (generalised estimating equation repeated-measures analysis). Six participants (19%) used an assistive device during the 6MWT at the phase 3 baseline, decreasing to three (10%) at phase 3 week 72a, and one from week 12b. BFI, Brief Fatigue Inventory; BL, baseline; BPI-SF, Brief Pain Inventory short-form; LSM, least squares mean; MCID, minimum clinically important difference; 6MWT, 6-Minute Walk Test; PRO, patient-reported outcome; WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index.
Ambulatory function
The mean 6MWT distance increased from 360 metres at baseline (range 197–569 metres) to 443 metres (341–574 metres) at the end of the open-label extension. The change from baseline was significant from week 24a through to the end of the open-label extension (figure 3F). Six participants (19%) used an assistive device during the 6MWT at the phase 3 baseline, decreasing to three (10%) at phase 3 week 72a and one from week 12b in the extension study (figure 3F).
Exploratory analysis of treatment interruption
Twenty-three participants received at least one dose of compassionate burosumab treatment between the two studies; of these, eight received continuous burosumab for 6–16 months and eight missed only one dose during 6–7 months’ treatment. Seven participants did not receive any compassionate burosumab treatment between studies (five were under the care of sites that did not participate in the programme, one was taking a treatment break during pregnancy and one declined treatment because they lived too far from the research site). These participants were without treatment for 8–15 months.
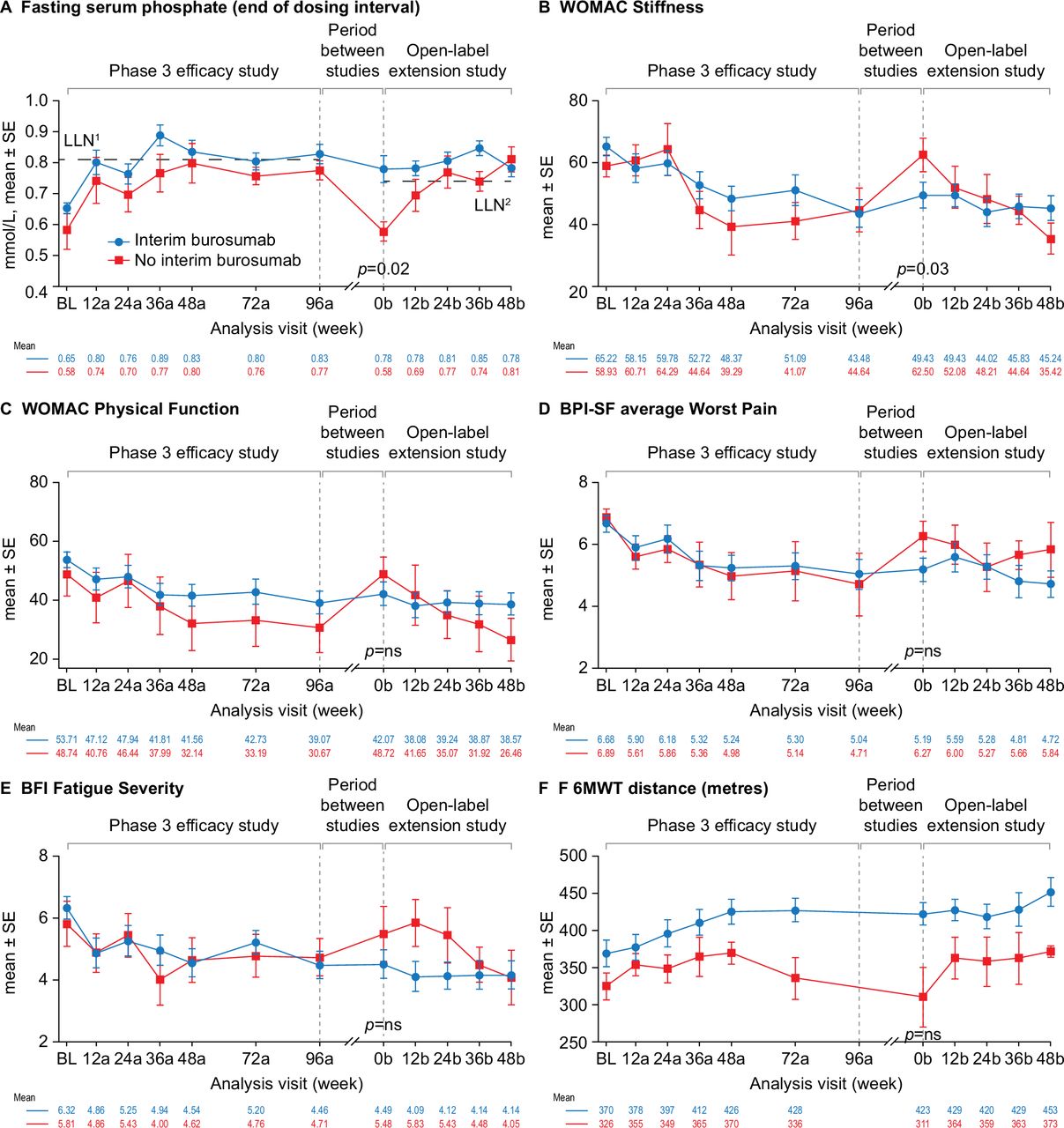
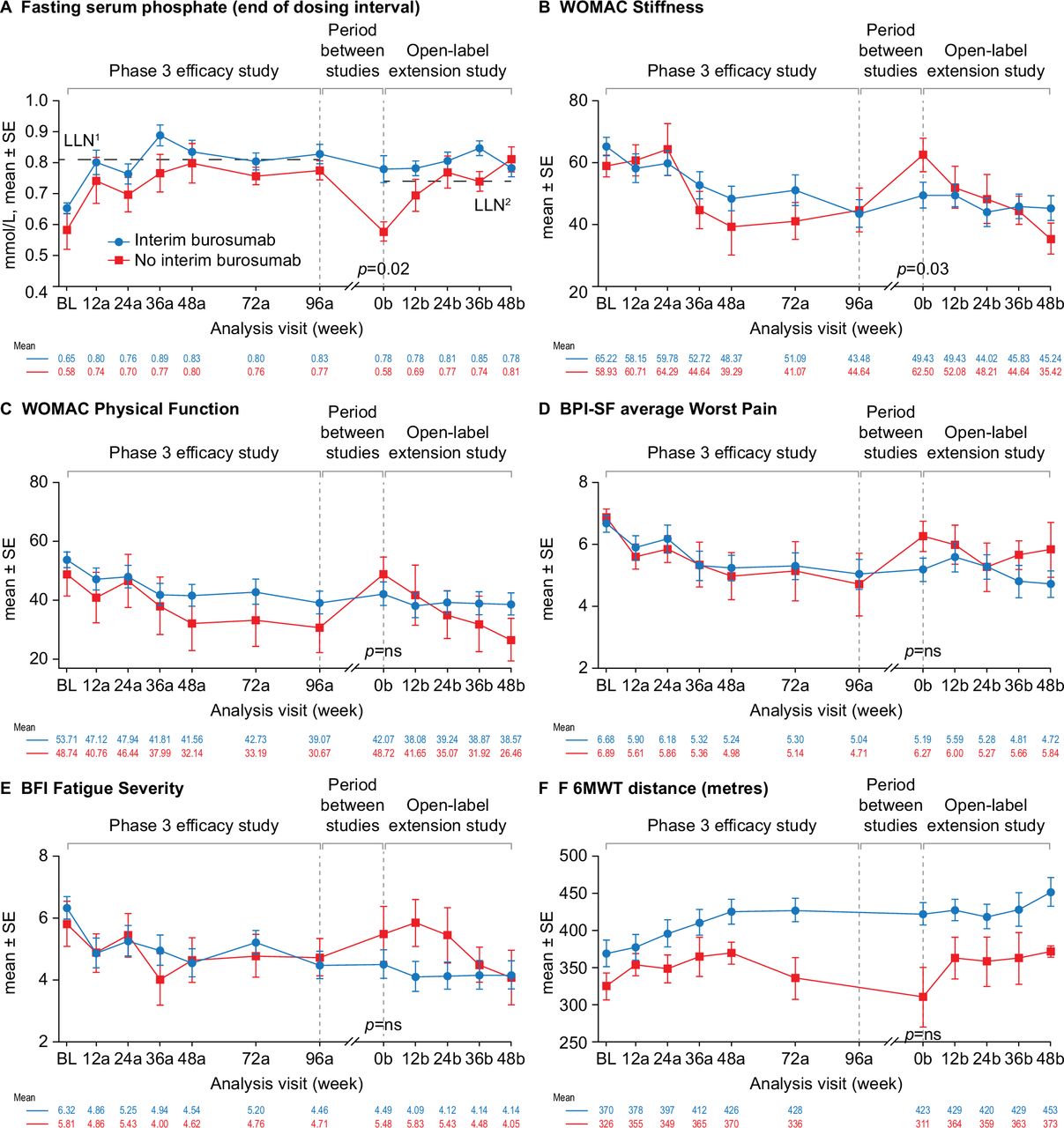
At entry into the open-label extension study (week 0b), fasting serum phosphate concentration was above the LLN in 52% of the participants (n=12) who received compassionate burosumab treatment between studies but in none of the participants who did not receive compassionate burosumab treatment (p=0.02) (figure 4A). Similar patterns were seen for TmP/GFR (29% (n=6) vs none) and serum 1,25(OH)2D concentrations (83% (n=19) vs 43% (n=3)) but these differences between the groups were not significant (online supplemental figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of burosumab treatment interruption on serum phosphate, PROs and 6MWT. Interim burosumab, n=23; no interim burosumab, n=7. Analysis weeks in the phase 3 study and open-label extension are indicated by ‘a’ and ‘b’ suffixes, respectively. A decrease in scores indicates improvement on the WOMAC, BPI-SF and BFI. An increase in distance on the 6MWT indicates improvement. BPI-SF and BFI data were captured at a single site visit and were not completed as part of a patient diary at weeks 72a and 96a. Fasting serum phosphate p values are for the difference between the groups (end of dosing cycle) at week 0b (tested using Fisher’s exact test); 52% of the interim burosumab group but none of no interim burosumab group had values ≥LLN at the start of the open-label extension period (p=0.01; Fisher’s exact test). PROs and 6MWT (tested using the Mann-Whitney U test) p<0.05 was considered significant. There was no significant difference between the groups at study baseline. Serum phosphate samples from the two studies were measured at different central laboratories, with different LLN values: 0.81 mmol/L in the phase 3 study (LLN1) and and 0.74 mmol/L in the open-label extension (LLN2). BFI, Brief Fatigue Inventory; BL, baseline; BPI-SF, Brief Pain Inventory short-form; LLN, lower limit of normal range; 6MWT, 6-Minute Walk Test; PRO, patient-reported outcome; WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index.
Improvements in WOMAC Stiffness and Physical Function scores, average BPI-SF Worst Pain scores, BFI Fatigue Severity scores and 6MWT distance during the phase 3 study were maintained at entry into the open-label extension study (week 0b) in participants who received compassionate burosumab treatment whereas scores returned to a similar level to phase 3 baseline in participants who did not receive compassionate burosumab (figure 4). The improvement from phase 3 baseline was significantly different between the two groups at the start of the open-label extension study for the WOMAC Stiffness score (3.6 vs −15.3; p=0.03) (figure 4B). Similar profiles were seen for the other PRO endpoints (online supplemental figure 5). The two treatment groups were not comparable at phase 3 baseline for 6MWT distance: the group that did not receive compassionate burosumab walked less far at baseline and at each visit in the phase 3 study compared with those who later received compassionate burosumab.
Discussion
This analysis demonstrates that the correction of serum phosphate concentrations to above the LLN observed in the phase 3 trial through to week 96 is maintained with long-term burosumab treatment for a further 48 weeks in the open-label extension. This is consistent with the mechanism of action of burosumab in increasing the absorption and reabsorption of phosphate, the latter demonstrated by the improvement in TmP/GFR. Serum 1,25(OH)2D concentrations were also maintained with long-term treatment.
The consistent burosumab dose through the phase 3 study reflects the requirement in the study protocol for the dose to remain fixed for the duration of the study, provided the serum phosphate concentration did not exceed 1.61 mmol/L at any time or 1.45 mmol/L on two occasions and body weight did not change by >20% from the baseline assessment. In the open-label extension study, several participants required protocol-permitted adjustments to the burosumab dose to maintain fasting serum phosphate within the normal range.
Consistent with the maintenance of serum phosphate levels, improvements in patient-reported pain, stiffness, physical function and fatigue, and ambulatory function (6MWT) were maintained with long-term burosumab treatment, alongside decreased use of pain medication, including opioids. Clinically meaningful improvements were consistently observed across most PRO endpoints from week 36a in the phase 3 study and maintained through to week 48b in the open-label extension study. The greatest improvements in ambulatory function were seen at later time points (ie, 6MWT distance at week 48b of the open-label extension) alongside less reliance on assistive devices.
The exploratory analysis suggests that the benefits of burosumab treatment on clinical laboratory tests of efficacy, PROs and ambulatory function are lost if treatment is interrupted, such as during the period between the two studies. Notably, in the group that did not receive compassionate burosumab treatment between studies (an interruption of 2 months or longer), serum phosphate levels returned to baseline levels measured before treatment. The changes in phosphate concentration with the interruption to and reinstatement of burosumab treatment are reflected in PROs and ambulatory function. However, there did not appear to be a rebound effect—clinical laboratory tests of efficacy, PROs and ambulatory function did not fall below the phase 3 study baseline, despite up to 15 months’ interruption to burosumab treatment. The analysis did not enable us to determine the impact of stopping treatment on clinical events such as a pseudofracture. Together, the observations indicate that continued treatment with burosumab is required to maintain serum phosphate within the normal range and thus maintain the benefits in mitigating pain, stiffness and fatigue and improving physical function and mobility. Notably, it took 36–48 weeks for the clinical benefits of treatment to return following interruption to treatment. This is consistent with the phase 3 study, in which the beneficial effect of treatment on PROs and ambulatory function improved up to about 36 weeks and stabilised thereafter.15 These findings indicate the need to examine the effects of continued treatment, and to determine whether interruptions to treatment, for example during pregnancy, affect long-term outcomes for patients, in order to inform treatment decisions.
The analysis reported here has some limitations. Participants were not randomised to receive or not receive compassionate burosumab treatment, so residual confounding cannot be excluded. While this analysis indicates that the clinical effect of burosumab wears off if treatment is interrupted, and returns with reinstatement of treatment, it would be reasonable to hypothesise that participants who received one compared with six doses over a 6-month period would have different outcomes during that period. Treatment during the interim period was not captured, so it is not known whether participants received conventional therapy during the interval. As this was a retrospective exploratory analysis and was not powered, interpretation is limited by small numbers of participants. In addition, while laboratory-specific normal ranges were used, samples were analysed by different central laboratories and results from the two studies cannot be compared directly. In addition, because of restrictions imposed during the COVID-19 pandemic, a small number of serum phosphate results were assessed at local laboratories and some data for the ambulatory assessment (6MWT) are missing at weeks 36b and 48b.
Data supporting the conclusion that burosumab is a safe long-term treatment for adults with XLH have been reported for participants receiving burosumab for an average of more than 3 years.24 The results reported here provide further safety evidence, with no grade 4 or 5 TEAEs reported and no treatment-emergent SAEs. Further safety data are being collected in a post-authorisation registry study of burosumab,25 26 which will provide important insight into the use of burosumab in a real-world setting.
In conclusion, the effect of burosumab treatment in restoring serum phosphate concentrations to within the normal range is maintained with continued treatment beyond 96 weeks, and also maintains benefits in associated clinical laboratory tests of efficacy, PROs and functional measures. Exploratory analysis indicates that these benefits are lost with interruptions to treatment but recover over time following reinstatement of treatment. Continued, uninterrupted burosumab therefore appears necessary to sustain the clinical and biological benefits of treatment.
Data availability statement
Data are available upon reasonable request. Data are available on reasonable request from the corresponding author. All data relevant to the study are included in the article or as online supplemental file 1. The study protocol and statistical analysis plan for this study will be available on the relevant clinical trial registry websites with the tabulated results.
Ethics statements
Patient consent for publication
Ethics approval
Both studies were designed, conducted, recorded, and reported in accordance with the principles established by the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. The Institutional Review board or Ethics Committee for each site approved the study protocols. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank the participants, patients and healthcare professionals who participated in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors A-LL, JSW, KB, MKJ, MC-S, MLB, PK, SHR, RK, RHL, RKC and SK were investigators on the study. AN and MN conducted the data analysis with oversight by WS. AN, AJR and AW drafted the initial manuscript content. All authors were involved in critically reviewing and revising the manuscript and providing final approval of the submitted version. PK is responsible for the overall content as guarantor.
Funding This study was funded by Ultragenyx Pharmaceutical and Kyowa Kirin International.
Competing interests The following authors served as clinical investigators for one or more studies, including this trial, sponsored by Ultragenyx Pharmaceutical in partnership with Kyowa Kirin International: A-LL, JSW, KB, MC-S, MKJ, MLB, PK, RKC, RHL, SHR and SK. A-LL, RK and RHL have received honoraria from Kyowa Kirin International for serving as an advisory board member. A-LL, RKC and RK have also received honoraria from Kyowa Kirin International for delivering presentations, and A-LL has received support from Kyowa Kirin International for attending meetings. MKJ and RHL have received consulting fees and grants from Kyowa Kirin International outside of the submitted work. SHR has received clinical trial funding from Amgen, UCB and AstraZeneca. AJR and AW are employees of Kyowa Kirin International and WS is an employee of Kyowa Kirin Pharmaceutical Development. AN and MN are employees of Chilli Consultancy and have received consultancy fees from Kyowa Kirin International to support the data analysis and medical writing of this manuscript and for projects outside this submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.