Article Text

Abstract
Objective To evaluate the long-term safety profile for upadacitinib across rheumatoid arthritis (RA), psoriatic arthritis (PsA), ankylosing spondylitis (AS) and atopic dermatitis (AD).
Methods Safety data from clinical trials of upadacitinib 15 mg and upadacitinib 30 mg (AD only) for treating RA, PsA, AS and AD as of 30 June 2021 were analysed; some RA and PsA studies included adalimumab and methotrexate as active comparators. Treatment-emergent adverse events (TEAEs) were presented by disease as exposure-adjusted event rates per 100 patient years (E/100 PY).
Results The analysis included 6991 patients (RA, n=3209; PsA, n=907; AS, n=182; AD, n=2693) who received at least one dose of upadacitinib, representing 15 425 PY of exposure (maximum duration 2.75–5.45 years) across diseases. Rates (E/100 PY) of any TEAE (205.5–278.1) and TEAE leading to discontinuation (4.5–5.4) were similar across diseases; serious TEAEs were numerically higher in patients with RA and PsA. Rates of herpes zoster (1.6–3.6), non-melanoma skin cancer (0–0.8) and elevations in creatine phosphokinase levels (4.4–7.9) were higher with upadacitinib than with active comparators in the RA and PsA populations. Deaths (0–0.8), serious infections (0–3.9), major adverse cardiovascular events (0–0.4), venous thromboembolism (<0.1–0.4) and malignancies (0.3–1.4) were observed, with rates generally lowest in AS and AD. Increased rates of acne were observed in patients with AD only.
Conclusions Findings from this analysis demonstrate that upadacitinib is generally well tolerated with observed differences in safety profiles likely reflective of varying patient characteristics across RA, PsA, AS and AD populations.
Trial registration numbers NCT02675426, NCT02706951, NCT02706847, NCT02629159, NCT02706873, NCT03086343, NCT03104374, NCT03104400, NCT03178487, NCT03569293, NCT03568318 and NCT03607422.
- Arthritis, Psoriatic
- Arthritis, Rheumatoid
- Spondylitis, Ankylosing
- Antirheumatic Agents
- Inflammation
Data availability statement
Data are available upon reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvieclinicaltrials.com/hcp/data-sharing/.html.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Upadacitinib is a Janus kinase (JAK) inhibitor with established efficacy in adults with rheumatoid arthritis (RA), psoriatic arthritis (PsA), ankylosing spondylitis (AS) and ulcerative colitis and adults and adolescents with atopic dermatitis (AD).
Several safety risks have been associated with JAK inhibitor use, including herpes zoster, serious and opportunistic infections, elevations in creatine phosphokinase (CPK) levels, major adverse cardiovascular events (MACEs), thromboembolic events and malignancies.
WHAT THIS STUDY ADDS
This integrated safety analysis of upadacitinib, based on more than 6000 patients and 15 000 patient-years of exposure across RA, PsA, AS and AD phase IIb/III trials, supports a reasonably acceptable safety profile for the treatment of patients with RA, PsA, AS and AD with no new safety risks compared with other JAK inhibitors.
Events of malignancy excluding non-melanoma skin cancer (NMSC), MACE and venous thromboembolism were observed at similar rates between upadacitinib and the active comparators adalimumab and methotrexate; known differences in the adverse event (AE) profile of JAK inhibitors, such as increased rates of herpes zoster, CPK elevations and NMSC, were observed.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The results of this integrated safety analysis of six trials in RA, two trials in PsA, one trial in AS and three trials in AD suggest that upadacitinib has a similar safety profile across RA, PsA, AS and AD with some variations in AE rates due to differences in patient population and disease-associated comorbidities.
Introduction
Immune-mediated inflammatory diseases (IMIDs), including rheumatoid arthritis (RA) and spondyloarthritides, are common, clinically diverse and chronic. While significant variations in population distribution, tissue localisation, clinical phenotypes and therapeutic responses are apparent between different IMIDs, they share many similar pathophysiological mechanisms.1–3 Small-molecule Janus kinase (JAK) inhibitors are a therapeutic class for the management of IMIDs.4 5 Although certain adverse events (AEs) appear associated with approved JAK inhibitors, including higher rates of herpes zoster and elevation in creatine phosphokinase (CPK) levels, the safety of individual JAK inhibitors continues to be evaluated in diverse patient populations impacted by each of these chronic conditions.3–8 Recent outcomes of the Oral Rheumatoid Arthritis triaL (ORAL) Surveillance study, comparing the JAK inhibitor tofacitinib to tumour necrosis factor (TNF) inhibitor therapy, further highlight the need to characterise the safety profile of JAK inhibitors, especially in the context of active comparators.9 10
Upadacitinib is an oral, reversible JAK inhibitor that has been and continues to be studied in comprehensive phase III clinical programmes including trials in RA, psoriatic arthritis (PsA), ankylosing spondylitis (AS), atopic dermatitis (AD) and ulcerative colitis (UC).11–22 While 15 mg once a day (QD) is the approved dose for treating rheumatological diseases, upadacitinib 15 and 30 mg (if response is inadequate with 15 mg) QD are approved for treating moderate-to-severe AD in adults and adolescents, and upadacitinib 45 mg was recently approved in the USA for treating patients with UC as induction therapy for 8 weeks followed by QD upadacitinib 15 mg or 30 mg as maintenance therapy.23 Here, we report an integrated analysis of the safety profile of QD upadacitinib across indications approved as of December 2021 including RA, PsA, AS and AD, along with active comparator data from trials including adalimumab or methotrexate (MTX).
Methods
Patients and studies
This analysis included safety data from 12 upadacitinib clinical trials (online supplemental table S1) with a cut-off date of 30 June 2021 for all studies. Studies of RA, PsA and AS included patients aged ≥18 years, while the studies of AD included adult and adolescent (aged ≥12 years) patients.11–21
Supplemental material
Patient involvement
Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Dosing
Data are pooled across studies within each disease and reported for the following treatment groups: upadacitinib 15 mg QD (RA, PsA, AS and AD), upadacitinib 30 mg QD (AD only, the only included indication approved at the 30 mg dose), adalimumab 40 mg subcutaneously every other week (RA [SELECT-COMPARE study], PsA [SELECT-PsA 1 study] and MTX [RA, SELECT-EARLY study]). Depending on study protocol, patients receiving upadacitinib did so either alone as monotherapy or in combination with MTX or other conventional synthetic disease-modifying antirheumatic drug (csDMARD) therapy. In SELECT-COMPARE, all patients, including those receiving upadacitinib and adalimumab, received background MTX; in SELECT-PsA 1, patients could have received adalimumab with or without concomitant background csDMARDs. All MTX data come from SELECT-EARLY (RA) and reflect monotherapy use.
Safety assessments
We report an overview of treatment-emergent adverse events (TEAEs) and adverse events of special interest (AESIs) to the JAK inhibitor class. TEAEs are defined as onset on or after the first dose of study drug and up to 30 days after the last dose of upadacitinib or MTX or up to 70 days after the last dose for adalimumab. Non-TEAEs are included only for deaths. An additional subanalysis is provided for TEAEs in patients receiving upadacitinib as monotherapy or in combination with any csDMARD at baseline for RA and PsA only.
TEAEs were coded using the Medical Dictionary for Regulatory Activities System Organ Classes and Preferred Terms. All deaths, potential cardiovascular (CV) events and arterial and venous thromboembolisms (VTEs) were adjudicated by a blinded, independent cardiovascular adjudication committee. Gastrointestinal perforations were blindly adjudicated by sponsor-employed experts independent of the upadacitinib clinical programmes using a prespecified definition of acute gastrointestinal tract perforation, which is non-iatrogenic and non-traumatic.
Major adverse cardiovascular events (MACEs) included CV death, non-fatal myocardial infarction and non-fatal stroke. VTE included deep vein thrombosis (DVT) and pulmonary embolism (PE). CV risk factors included history of a CV event, hypertension or diabetes mellitus; current or former tobacco use; elevated low-density lipoprotein cholesterol (≥3.36 mmol/L) levels or lowered high-density lipoprotein cholesterol (<1.034 mmol/L) levels. Active tuberculosis and herpes zoster are separately assessed from other opportunistic infections. Laboratory-related abnormalities (anaemia, neutropenia, lymphopenia, hepatic disorder and elevated CPK levels) are based on investigator-reported AEs. Potentially clinically significant values (grade 2, 3 and 4 changes) for select laboratory parameters of interest to the JAK inhibitors class are also provided. For RA, the toxicity grading scales are based on OMERACT (Outcome Measures in Rheumatoid Arthritis Clinical Trials) criteria, with the exception of creatine kinase and creatinine, which are based on NCI Common Terminology Criteria for Adverse Events (CTCAE). For PsA, AS and AD, all toxicity grading scales are based on CTCAE.
Statistical analyses
All TEAEs are reported as exposure-adjusted event rates (EAERs, exposure-adjusted event rates per 100 patient years [E/100 PY]) based on the treatment received at the time of the AE with 95% CIs calculated using the exact method for the Poisson mean; all events, including multiple events occurring in a single patient, were included in the numerator. For EAERs, exposure time was calculated as the total study drug duration. We also calculated exposure-adjusted incidence rates (EAIRs) with 95% CIs using the exact method for the Poisson mean for AESIs (online supplemental material); EAIRs were calculated as the number of patients with one or more event per 100 PY (n/100 PY). In patients who experienced an event, exposure time was calculated as the time to the first event; in patients who did not experience an event, exposure time was censored on the day of the patient’s last assessment or cut-off date of database lock, whichever occurred first. Additional subanalyses include EAER stratified by 6-month intervals (reported for MACE, VTE, malignancy excluding non-melanoma skin cancer (NMSC), serious infections and herpes zoster) and EAERs for all AESIs stratified by age group (patients aged <65 years and ≥65 years).
Standardised mortality ratio (SMR) was determined using the WHO country-specific, age-specific and gender-specific death data for the general population; 95% CIs were calculated using Byars’ approximation, and calculations included COVID-19 deaths. Standard incidence ratio (SIR) for malignancy excluding NMSC was determined using age-gender-adjusted data from the Surveillance, Epidemiology, and End Results (SEER) cancer age-specific and gender-specific incidence rate data in 2000–2018 from the US general population and adjusted for age at baseline; 95% CIs were calculated following a Poisson distribution.
Results
Patients and exposure
Patient data collected from 12 studies included 3209 patients with RA (PY=9079.1), 907 with PsA (PY=1872.3), 182 with AS (PY=320.1) and 2693 with AD (PY=2035.8 for upadacitinib 15 mg and 2118.0 for upadacitinib 30 mg). The maximum duration of treatment was longest in the RA programme (5.45 years, median 3.46 years) and shortest in the AD programme (2.75 years, median 1.62 years). Reference comparator arms included patients receiving at least one dose of adalimumab (RA: 579 patients, 1307.7 PY; PsA: 429 patients, 903.7 PY) and patients receiving at least one dose of MTX (314 patients with RA only, 781.7 PY). The majority of patients receiving upadacitinib also received concomitant csDMARD therapy in RA (2548 patients) and PsA (642 patients), whereas concomitant csDMARD use was infrequent in patients with AS and AD (table 1) . Other noteworthy differences in baseline characteristics typical of the respective disease state were present, including younger patients in the AD population and a higher proportion of female patients in the RA population (table 1). Across all groups, 48.5%–83.5% of patients had at least one CV risk factor at baseline.
Baseline demographics and disease characteristics
Overview of AEs
Rates of any TEAEs with upadacitinib ranged from 205.5 with upadacitinib 15 mg in RA to 278.1 with upadacitinib 30 mg in AD (table 2). In RA, the rates were generally comparable between upadacitinib and active comparators and in patients receiving upadacitinib as monotherapy or in combination with csDMARD therapy (online supplemental table S2); in PsA, the rate was numerically higher with upadacitinib (244.8) vs adalimumab (229.9). The highest rates of serious TEAEs were observed in RA. TEAEs leading to discontinuation were generally comparable across all treatment groups and diseases. In AD, rates of any TEAE, serious TEAE and TEAE leading to discontinuation were numerically higher with upadacitinib 30 mg compared with upadacitinib 15 mg. The incidence of deaths was <1.0/100 PY in the RA programme and similar across upadacitinib, adalimumab and MTX groups. In PsA, rates of death were higher with upadacitinib 15 mg compared with adalimumab, owing to increased COVID-19-related deaths in patients taking upadacitinib. No deaths were reported in AS, and three deaths were reported in the upadacitinib 30 mg AD group, with two of the three deaths related to COVID-19. Excluding COVID-19, the most common cause of death was related to CV disease, followed by other infections and malignancies. The SMR estimates for each disease were 0.59 (95% CI: 0.43 to 0.78) for RA, 0.59 (95% CI: 0.36 to 0.92) for PsA and 0.28 (95% CI: 0.06, 0.82) for AD (both doses), with no evidence suggesting that the number of deaths in patients with RA, PsA or AD exposed to upadacitinib were higher than what would have been expected for the general population.
Exposure and overview of TEAEs
In RA, PsA, AS and AD populations, the most common TEAE associated with upadacitinib 15 mg was upper respiratory tract-related infection (online supplemental table S3), while acne was the most common TEAE associated with upadacitinib 30 mg in AD. Acne was infrequently reported in the rheumatological diseases; while events of acne in AD were generally mild or moderate, non-serious and rarely led to treatment discontinuation. The pattern, characteristics and incidence of COVID-19 infections, including frequency of events and those leading to hospitalisation, observed in patients receiving upadacitinib were generally similar to what has been observed in the general population (online supplemental table S4).
Adverse events of special interest
AESIs are presented in figure 1 in EAERs and in the online supplemental figure S1 in EAIRs.
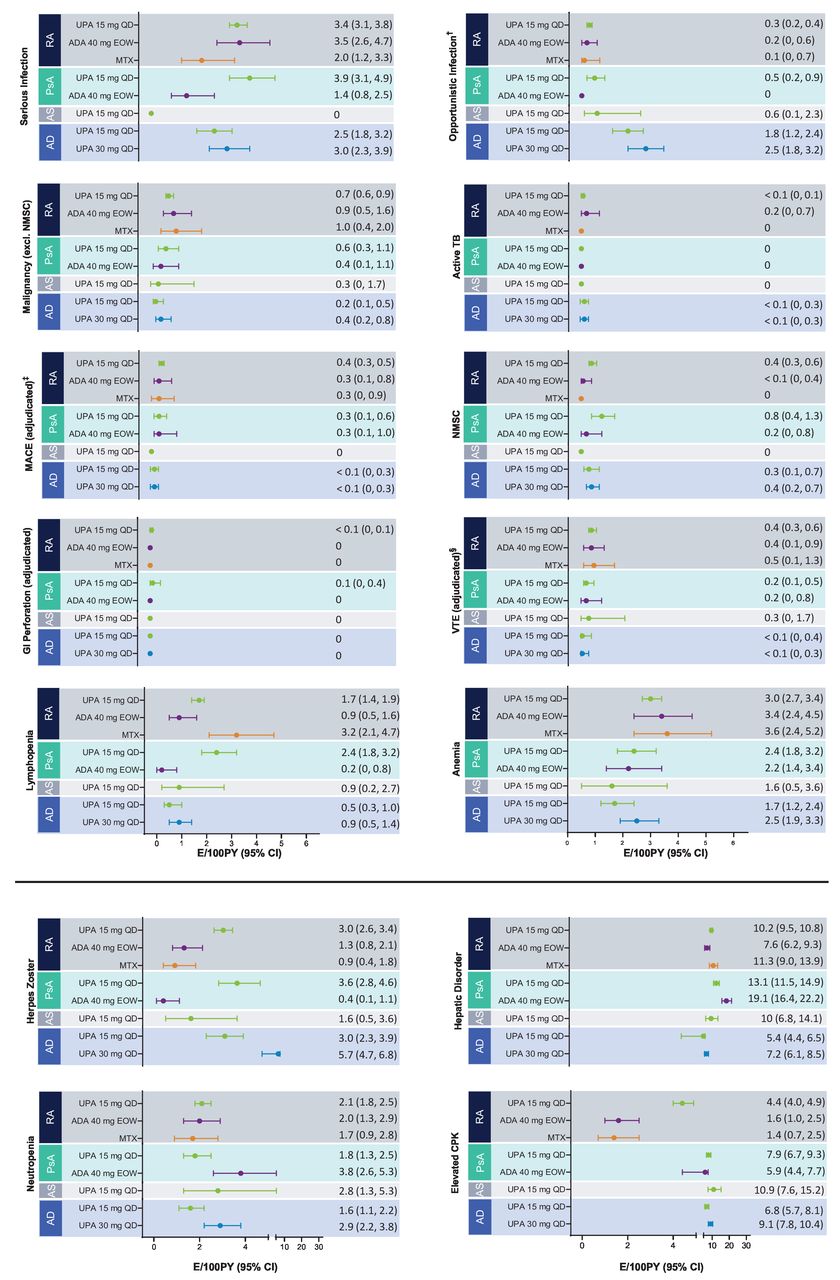
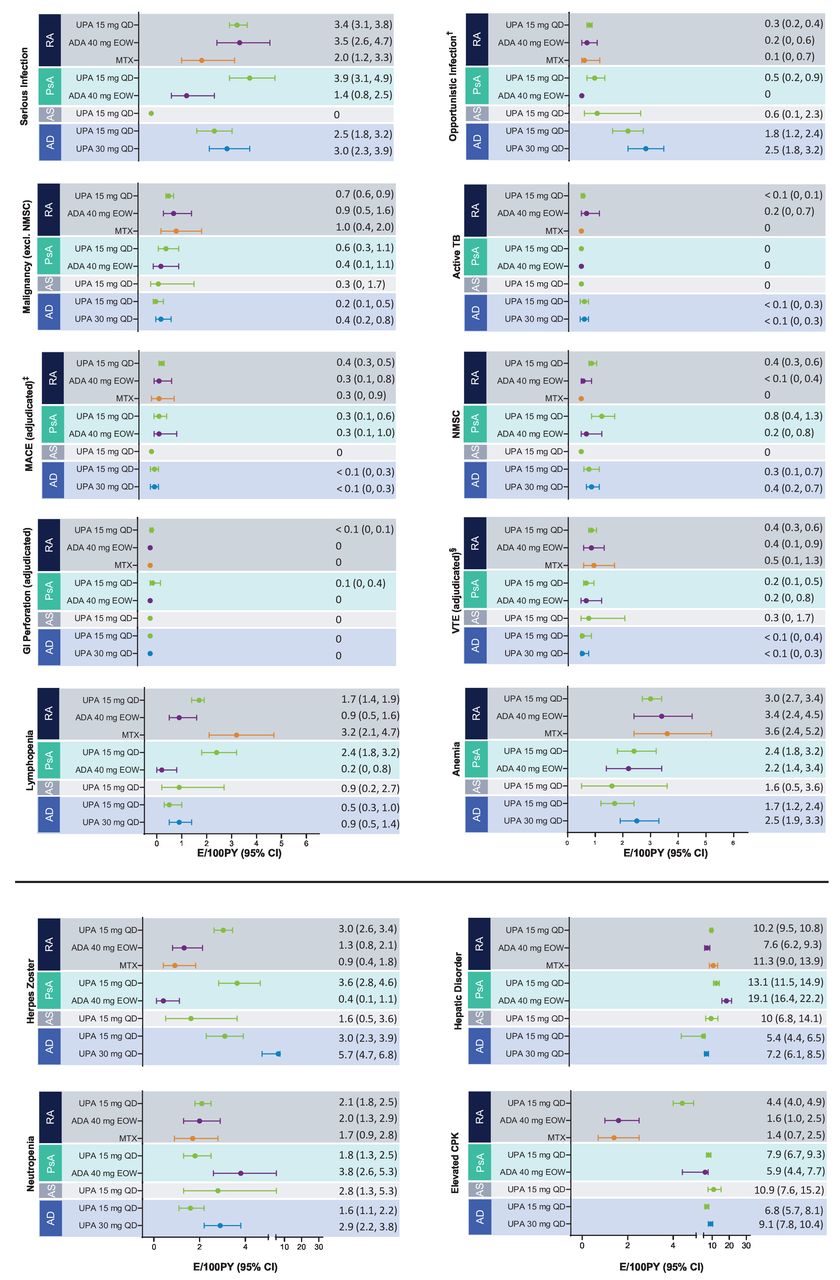{kind=link}
Exposure-adjusted event rates for TEAEs of special interest.* †Excluding TB, oral candidiasis and herpes zoster. ‡Defined as cardiovascular death, non-fatal myocardial infarction and non-fatal stroke. §Including deep vein thrombosis and pulmonary embolism. *RA: UPA 15 mg QD (n=3209), ADA 40 mg EOW (n=579), MTX (n=314); PsA: UPA 15 mg QD (n=907), ADA 40 mg EOW (n=429); AS: UPA 15 mg QD (n=182); AD: UPA 15 mg QD (n=1340), UPA 30 mg QD (n=1353). AD, atopic dermatitis; ADA, adalimumab; AS, ankylosing spondylitis; CPK, creatine phosphokinase; E, event; EOW, every other week; GI, gastrointestinal; MACE, major adverse cardiovascular event; MTX, methotrexate; NMSC, non-melanoma skin cancer; PsA, psoriatic arthritis; PY, patient years; QD, once a day; RA, rheumatoid arthritis; TB, tuberculosis; TEAE, treatment-emergent adverse event; UPA, upadacitinib; VTE, venous thromboembolic event.
Serious and opportunistic infections
Serious infections with upadacitinib occurred at similar rates between RA, PsA and AD and infrequently led to discontinuation; no serious infections were reported in AS. In RA, serious infections with upadacitinib 15 mg were reported at rates similar to those reported for adalimumab, both of which had higher rates than MTX. The most common serious infection reported with upadacitinib treatment across all disease states was COVID-19 followed by pneumonia for RA and PsA, and herpes zoster for AD. Within each disease state, the rates of serious infections were relatively stable over time (online supplemental figure S2). Increased rates of serious infection in PsA observed ≥24 months appear to have been driven by events of COVID-19, reflecting the timing of these studies relative to that of the pandemic. Overall rates of serious infections including and excluding COVID-19 were 3.4 vs 2.8 E/100 PY for RA, 3.9 vs 2.1 E/100 PY for PsA, 2.5 vs 2.2 E/100 PY for upadacitinib 15 mg in AD and 3.0 vs 2.3 E/100 PY for upadacitinib 30 mg in AD.
Opportunistic infections were infrequently reported in all disease states, with highest rates observed in AD. In rheumatological diseases, the most commonly reported opportunistic infection was oesophageal candidiasis (online supplemental table S5). In AD, the rate of opportunistic infection (excluding herpes zoster and tuberculosis) was numerically higher with upadacitinib 30 mg compared with upadacitinib 15 mg (2.5 and 1.8 E/100 PY, respectively); almost all events were reported as eczema herpeticum or the synonymous Kaposi’s varicelliform eruption.
Herpes zoster
Rates of herpes zoster among patients receiving upadacitinib were similar across diseases, occurring in a dose-dependent manner and more frequently than with adalimumab and MTX. No apparent correlation between length of upadacitinib 15 mg exposure and herpes zoster rates was observed across RA, PsA, AS or AD, although an upward trend with longer exposure to upadacitinib 30 mg was observed in AD (online supplemental figure S3). The majority of herpes zoster events with upadacitinib were mild or moderate, infrequently led to discontinuation and involved a single dermatome (76% in RA, 67% in PsA, 100% in AS, and 78% for upadacitinib 15 mg and 67% for upadacitinib 30 mg in AD), with no events involving the central nervous system or internal organs (online supplemental table S6). Patients were permitted to restart upadacitinib therapy on resolution of a herpes zoster episode.
Malignancies
Malignancy excluding NMSC
Malignancies (excluding NMSC) were reported in all disease states with rates ≤1.0 E/100 PY overall, appearing consistent across diseases and between upadacitinib and active comparators. In AD, while malignancy rates were numerically higher with upadacitinib 30 mg vs upadacitinib 15 mg, four of nine malignancy events reported in the upadacitinib 30 mg group were diagnosed within 6 months after the first dose of upadacitinib. There was no significant change in rate of malignancies excluding NMSC over time of exposure to upadacitinib across diseases and doses (online supplemental figure S4). No pattern or cluster of malignancies excluding NMSC was observed in RA, PsA and AD (online supplemental table S7). Only one malignancy excluding NMSC of stage IVa squamous cell carcinoma of the tongue was observed in the AS study; the patient was a former smoker with less than 5 months of drug exposure. The age–gender adjusted SIR for malignancies other than NMSC using malignancy data from the SEER 18 Registry Research Data 2000–2018 for the general population was estimated at 1.00 (95% CI 0.80 to 1.24) for the upadacitinib 15 mg group in RA, 0.81 (95% CI 0.41 to 1.46) in PsA and 0.50 (95% CI 0.10 to 1.47) in AD; the ratio for patients receiving upadacitinib 30 mg in AD was 1.10 (95% CI 0.53 to 2.03).
Non-melanoma skin cancer
Rates of NMSC (≤0.8 E/100 PY) were generally consistent across diseases for upadacitinib with no events observed in AS. However, event rates of NMSC were numerically higher with upadacitinib compared with adalimumab and no events were reported with MTX. Slightly higher rates of NMSC were also observed in the upadacitinib 30 mg group vs the 15 mg group in AD. Events of NMSC were generally non-serious and did not lead to treatment discontinuation.
Major adverse cardiovascular event
MACE was reported across all treatment groups with rates of <0.5/100 PY, and no events were observed with upadacitinib 15 mg in AS. Rates of MACE were comparable between upadacitinib, adalimumab and MTX in RA and PsA (0.3 E/100 PY–0.4 E/100 PY). One event of MACE (<0.1 E/100 PY) was reported each in the upadacitinib 15 mg and 30 mg groups of the AD programme. There was no observed relationship between time of exposure to upadacitinib and MACE (online supplemental figure S5). Most patients experiencing MACE had at least one CV risk factor. There were 11 CV deaths, all within the RA population.
Venous thromboembolism
VTE was observed among patients receiving upadacitinib across disease states with rates of 0.4 E/100 PY in RA, 0.2 E/100 PY in PsA, 0.3 E/100 PY in AS, and <0.1 E/100 PY for both upadacitinib 15 mg and 30 mg in AD. Rates with upadacitinib were comparable to those observed with adalimumab (RA and PsA) and MTX (RA). There was no relationship observed between time of exposure to upadacitinib and VTE (online supplemental figure S6). Events of PE were more frequent than DVT in RA, PsA and AS; in AD, PE and/or DVT, rates were <0.1 E/100 PY each for upadacitinib 15 mg and upadacitinib 30 mg. Most patients experiencing a VTE had at least one CV and/or thromboembolic risk factor. There were two fatal VTE events across all diseases, both being PEs in patients with RA.
Laboratory abnormalitiess
AEs of elevations in blood CPK levels occurred in all diseases, were dose dependent, occurred more frequently with upadacitinib than with adalimumab and MTX and were primarily asymptomatic. Few patients had symptoms of muscle pain, and alternative aetiologies of vigorous physical activity were identified in most cases. One case of rhabdomyolysis occurred in an adolescent patient receiving upadacitinib 30 mg for AD following a boxing activity. AEs of anaemia, neutropenia and lymphopenia were also reported in RA, PsA, AS and AD. Most laboratory abnormalities were reported to be mild overall and transient and few resulted in discontinuation of the study drug (these abnormalities are further discussed in the online supplemental material).
AEs related to laboratory parameters of special interest for the JAK inhibitor class (anaemia, lymphopenia, neutropenia, hepatic disorders and CPK elevations) are further discussed in the online supplemental material 1; abnormal laboratory results of haematology and chemistry tests by grading criteria performed during the upadacitinib studies are described in online supplemental tables S8 and S9.
AESIs by age
While numbers of patients aged ≥65 years are limited overall, event rates tended to be higher for MACE, VTE, malignancies and serious infections, regardless of treatment (upadacitinib, adalimumab or MTX; online supplemental table S10). In RA and PsA, rates of these events in patients aged ≥65 years were similar between upadacitinib 15 mg and adalimumab.
Additional information on AESIs by concomitant therapy
Overall, the event rates of AESIs occurring in patients receiving upadacitinib monotherapy or upadacitinib combination therapy with a csDMARD in RA and PsA were generally consistent (online supplemental figure S7).
Discussion
This safety analysis of 6991 patients receiving upadacitinib with a maximum of 5.45 years of follow-up appears largely consistent across RA, PsA, AS and AD for key AESIs such as MACE, VTE and malignancy excluding NMSC, and no new or unexpected safety risks were identified compared with previous reports.24–27 Differences in AE types and rates across disease states reflect expected distinctions between disease-specific patient populations and background risks.28–31 Long-term exposures with adalimumab (RA and PsA) and MTX (RA only), although with lower numbers of patients evaluated, within the upadacitinib clinical trial programme further enabled the contextualisation of the described upadacitinib safety profile alongside active comparators.
Varied comorbidities and patient populations across diseases likely resulted in differences in some TEAE types and rates.28–31 Notably, acne was one of the most common TEAEs within the AD population while minimally represented across others, possibly owing to a younger overall population of patients with AD or greater scrutiny of dermatological AEs by dermatologists. Acne has been observed in all trials of JAK inhibitors in AD, though most cases are mild to moderate and do not result in treatment discontinuation.32 Additionally, baseline concomitant corticosteroid use was higher among patients with RA, largely reflecting differences in treatment recommendations and guidelines; however, the relationship between concomitant corticosteroid use and events of MACE or VTE have not yet been explored.33–35 Determining a relationship between corticosteroid use and MACE or VTE is beyond the scope of this analysis, given the challenge of separating corticosteroid use from other risk factors, the need for further data regarding dosing (mean, median and cumulative) and given the included data concerns corticosteroid use at baseline only. The event rates of herpes zoster, NMSC and elevation in CPK levels were higher with upadacitinib compared with adalimumab and MTX in RA and/or PsA; however, the rates of the remaining AESIs varied without consistent findings between upadacitinib vs the active comparators in the two programmes. An increased risk of herpes zoster and elevated CPK levels is associated with JAK inhibition and consistent with the overall safety profiles of JAK inhibitors.8 24 36 37 Herpes zoster infections observed with upadacitinib were largely non-serious and limited to one dermatome, while elevations in CPK levels were mostly asymptomatic and transient. Of note, the upadacitinib label recommends herpes zoster vaccination for all patients prior to initiating upadacitinib therapy. Furthermore, there is a recognised potential for JAK inhibitors to be associated with an increased risk of NMSC, with systematic reviews of the use of tofacitinib, ruxolitinib and baricitinib identifying higher rates of NMSC in patients with IMIDs.38–40 Rates of NMSC were higher in patients receiving upadacitinib than active comparators, primarily presenting as basal or squamous cell carcinoma in patients aged ≥65 years. Further studies with more long-term data are required to fully determine any association between JAK inhibitors and NMSC. AESIs were similar across all diseases, except for an increase in opportunistic infections excluding herpes zoster in AD due to eczema herpeticum, which is commonly associated with AD.41An overall increased risk of TEAEs was observed in the upadacitinib 30 mg population compared with upadacitinib 15 mg for AD. COVID-19 infections had a notable impact on serious infection rates, especially in PsA. The timing of the PsA studies coinciding with that of the COVID-19 pandemic likely contributed, and when COVID-19 infections were excluded, there was little variation in serious infections with upadacitinib across diseases.
Recent data from ORAL Surveillance, an event-driven, head-to-head study in an RA population enriched for CV risk, demonstrated that tofacitinib failed to meet the non-inferiority criteria for events of MACE and malignancy (excluding NMSC) vs TNF inhibitor therapy.10 Patients most at risk of experiencing events of MACE or malignancy while receiving tofacitinib compared with TNF inhibitors were aged ≥65 years and had a history of smoking, underscoring the need to evaluate patient medical history when selecting therapeutic options.10 42 Of note, a similar signal to that observed in ORAL Surveillance has not been observed in the overall tofacitinib trial programme, and the separation between tofacitinib and TNF inhibitors only became apparent in a patient population enriched for overall risk. A definitive safety outcomes study like ORAL Surveillance has not been conducted with upadacitinib and whether the potential increased risk of these events reflects a JAK inhibitor class effect remains unclear.
Events of malignancies excluding NMSC were reported in patients receiving upadacitinib and were consistent with active comparators, with age-adjusted SIR data suggestive of no increased risk of these events compared with the general population. Numerically higher rates of malignancy were observed with upadacitinib 30 mg compared with upadacitinib 15 mg in AD; however, four of the nine malignancies observed with upadacitinib 30 mg occurred within 6 months after initiating upadacitinib.
VTE risk with JAK inhibitor therapy has become a concern owing to elevated rates observed with 4 mg baricitinib compared with placebo in several studies.6 7 A higher rate of VTE was also observed with tofacitinib 10 mg given two times a day compared with TNF inhibitors in ORAL Surveillance.10 Although events of VTE were reported in patients receiving upadacitinib across diseases, the rates appeared consistent with those observed for adalimumab in RA and PsA and MTX in RA and were consistent with background rates for the individual diseases.43 44 Several risk factors for VTE are inherently present in IMIDs including systemic inflammation, and patients with RA are at a substantially increased risk of VTE compared with the general population.44–46 The potential association between JAK inhibitors and VTE risk requires further analysis, given the inherent risk carried by the conditions for which JAK inhibitors are indicated.
Limitations of this analysis include a lack of extended placebo control arms for comparative analysis and lack of postmarketing data. Additionally, the pooling strategy for upadacitinib leveraged data sets which did not include active comparator arms. Substantial exposure time is represented; however, data for certain populations (AS) and active comparators are more limited and occurrence of certain events was low. Consequently, rate estimations for events such as malignancy, MACE and VTE have limited precision for conclusive interpretations. Additional follow-up is required to fully characterise the rates observed on upadacitinib for these and other long latency events. All reported TEAEs were reported in the closely monitored clinical trial setting, and the full safety profile of upadacitinib should also consider reported TEAEs from clinical practice settings and registries. Finally, only indications with approval as of December 2021 were included in this analysis, and, as such, UC was not included, thus limiting the further characterisations of events that may have a dose response.
Conclusions
Analyses of long-term data demonstrate that upadacitinib was generally well tolerated in RA, PsA, AS and AD with no new safety risks identified compared with previous reports. Some variations in events were observed across diseases, possibly reflecting differences in patient populations and disease-associated comorbidities that impact background risk. Follow-up of patients receiving upadacitinib will continue as these trials are ongoing.
Data availability statement
Data are available upon reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvieclinicaltrials.com/hcp/data-sharing/.html.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and is an integrated analysis of a clinical trials programme. All trials were conducted according to the International Conference on Harmonisation guidelines, the Declaration of Helsinki principles and applicable local country regulations. All study-related documents were approved by independent ethics committees and institutional review boards at each site. All patients provided written informed consent.
Acknowledgments
AbbVie and the authors thank the trial investigators and patients who participated in these clinical trials. Medical writing support was provided by Benjamin Holmes, DVM, and Kersten Reich, MPH, CMPP, of JB Ashtin, and was funded by AbbVie.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @drpnash
Contributors All authors had access to the data and participated in the development, review, critique and approval of the manuscript throughout the editorial process and approved the final manuscript draft submitted for publication. All authors agreed to be accountable for all aspects of the work, ensuring the accuracy and integrity of the publication. All named authors meet the ICMJE criteria for authorship for this article, take responsibility for the integrity of the work and have given their approval for this version to be published. Research project: (1) study concept and design: all authors; (2) data acquisition: GRB, SBC, PN, ADI, EM, YT and PJM; (3) statistical analysis: JL and HP; and (4) data interpretation: GRB, SBC, KLW, PN, ADI, AD, EM, YT, JL, APL, HP, TS, PJM and EG-Y. Manuscript review and critique: All authors. Manuscript guarantor: GRB.
Funding AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
Competing interests GRB received consulting fees and honoraria for lectures from AbbVie, Eli Lilly, Gilead and Pfizer and is editor of RMD Open. SBC received grants and consultation fees from Amgen, AbbVie, Boehringer Ingelheim, Gilead, Pfizer, Roche and Sandoz. KLW received consulting fees and/or research grants from AbbVie, BMS, Lilly, Pfizer, Roche Gilead, Galapagos and UCB. PN received research grants and consulting fees from AbbVie, Bristol-Myers Squibb, Celgene, Eli Lilly, Gilead and Janssen, and is a member of speakers’ bureaus for AbbVie, Bristol-Myers Squibb, Celgene, Eli Lilly, Gilead and Janssen. ADI received honorarium for consultancy from AbbVie, Arena Pharmaceuticals, Aslan, BenevolentAI, Chugai, Dermavant, Genentech, LEO Pharma, Lilly, Menlo Therapeutics, Novartis, Pfizer, Regeneron, Sanofi and UCB. AD received grant/research support from AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Novartis, Pfizer and UCB, and is a consultant for AbbVie, Amgen, Aurinia, Bristol Myers Squibb, Celgene, Eli Lilly, Glaxo Smith Kline, Janssen, MoonLake, Novartis, Pfizer and UCB. EM received or has pending grants from Roche, Pfizer, Bristol-Myers Squibb and Novartis. He had received honorarium from Eli Lilly, Pfizer, GlaxoSmithKline, Roche, Sanofi, AstraZeneca, Sandoz, Amgen, Gemmene and AbbVie; provided writing assistance, medicines, equipment or administrative support to Pfizer, AbbVie and Roche; and received payment for lectures including service on speakers’ bureaus from Eli Lilly, Pfizer, GlaxoSmithKline, Roche, Sanofi, AstraZeneca, Sandoz, Amgen, Gema Biotech and AbbVie. YT received speaking fees and/or honoraria from Daiichi-Sankyo, Eli Lilly, Novartis, YL Biologics, Bristol-Myers Squibb, Eisai, Chugai, AbbVie, Astellas, Pfizer, Sanofi, Asahi Kasei, GlaxoSmithKline, Mitsubishi-Tanabe, Gilead and Janssen, and research grants from Mitsubishi-Tanabe, Chugai, AbbVie, Takeda, UCB, Daiichi-Sankyo and Eisai. PJM received grants/research support from AbbVie, Amgen, Bristol-Myers Squibb, Inmagene, Janssen, Eli Lilly, Novartis, Pfizer and UCB; is a consultant for AbbVie, Acelyrin, Aclaris, Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Galapagos, Gilead, GlaxoSmithKline, Inmagene, Janssen, MoonLake, Novartis, Pfizer, Sun Pharma, INC Pharma and UCB; and is on speakers’ bureaus for AbbVie, Amgen, Janssen, Novartis, Pfizer, UCB and Crescendo Bioscience. EG-Y is an employee of Mount Sinai and received research funds (grants paid to her institution) from AbbVie, Almirall, Amgen, AnaptysBio, Asana Biosciences, AstraZeneca, Boehringer Ingelheim, Cara Therapeutics, Celgene, Eli Lilly, Galderma, Glenmark/Ichnos Sciences, Innovaderm, Janssen, KAO, Kiniksa, Kyowa Kirin, Leo Pharma, Novan, Novartis, Pfizer, Ralexar, Regeneron Pharmaceuticals and UCB; is a consultant for AbbVie, Almirall, Amgen, Arena, Asana Biosciences, Aslan Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Bristol-Meyers Squibb, Cara Therapeutics, Celgene, Connect Pharma, Eli Lilly, EMD Serono, Evidera, Galderma, Ichnos Sciences, Incyte, Janssen Biotech, Kyowa Kirin, Leo Pharma, Pandion Therapeutics, Pfizer, RAPT Therapeutics, Regeneron Pharmaceuticals, Sanofi, SATO Pharmaceutical, Siolta Therapeutics, Target Pharma Solutions, UCB and Ventyx Biosciences. JL, APL, HP and TS are full-time employees of AbbVie and may hold AbbVie stock or stock options.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.