Article Text

Abstract
Modern technological innovations have advanced our understanding of the genetic basis of spondyloarthritis. In ankylosing spondylitis (AS), where the major histocompatibility complex (MHC) accounts for nearly half of the predisposition, most comes from HLA-B27, for which 65 subtypes are now recognised, although other genes are also at work including HLA-B60 (B*40:01). Other genes have been identified, including those involved in peptide editing for loading onto class I MHC molecules (ERAP1) and cytokine genes such as interleukin 1A (IL-1A) and those involved in the Th17 network (IL-23R, an association seen primarily in Caucasians) and others. In acute anterior uveitis, these associations are also seen as well as a region on chromosome 9p and genes whose confirmation is under way. Psoriasis and psoriatic arthritis fall into this disease spectrum, with the largest region of susceptibility coming from the MHC (most likely HLA-C, ie, C*06:02 although additional influences are also being implicated), and most of the other genetic susceptibility coming from genes involved in cytokine production, specifically genes in the Th17 pathway (IL-12B, IL-23A and IL-23R, the latter, like in AS, not seen in Asians), genes in the nuclear factor κB pathway (TNFAIP3 and TNIP1) and genes in the Th2 pathway (IL-4 and IL-13). Given that more than half of patients with AS have evidence on colonoscopy of at least occult inflammatory bowel disease (IBD), it is not surprising that shared genetic influences are operative. In IBD, genes important in the innate immune response (NOD2), autophagy (ATG6L1) and regulation of the IL-23 pathway (IL-23R) play a role in disease susceptibility.
Statistics from Altmetric.com
Introduction
Recent years have witnessed rapid advances in our understanding of the genetic basis of spondyloarthritis and related diseases (ankylosing spondylitis (AS), acute anterior uveitis (AAU), psoriasis and psoriatic arthritis (PsA) and inflammatory bowel disease (IBD)). From HLA and other candidate gene studies, genetic methodologies have progressed through family studies with microsatellite marker to more recent dense explorations of the genome through computer-based gene chip technologies. This review will explore the more recent findings in spondyloarthritis and related disease susceptibility.
Genetics of ankylosing spondylitis
Studies of disease concordance in twins and families of patients with AS indicate that the susceptibility to the disease is largely due to genetic factors and that several loci are likely to be involved. Twin studies have shown that the MZ twin concordance rate is 63% compared with the DZ concordance rate of 12.5%.1 Genetic variance modelling has indicated that genetic factors contribute over 90% to overall susceptibility, with the rest due to random environmental effects.2 The environmental trigger(s) are therefore likely to be widespread (such as gut flora) and do not contribute significantly to the overall causation of the disease.
Role of HLA-B27
For nearly 40 years it has been known HLA-B27 formed the most significant association with AS, as well as with other types of spondyloarthritis (SpA), especially reactive arthritis (70% in white people), psoriatic spondylitis (60–70%), the spondylitis associated with IBD (50–60%) and AAU (50%). Other types of SpA show a much lower association (eg, peripheral PsA about 25% compared with a frequency of 6–8% in controls) or no association at all (eg, peripheral enteropathic arthritis).3
Over 69 subtypes of HLA-B27 have now been recognised,4 whose evolution from the parent B27 allele B*27:05 has largely followed geographical grounds reflecting human migrations (table 1). The overwhelming majority of HLA-B27 alleles in Western European Caucasians comprise HLA-B*27:05 (about 90%) and HLA-B*27:02 (about 10%). In patients from Eastern Asia, HLA-B*27:04 and B*27:05 are most commonly encountered, especially in patients with AS, and in Southern Asia HLA-B*27:07 is found. Two subtypes—HLA-B*27:06 (encountered in South-east Asia) and B*27:09 (found largely in Sardinia)—appear to have lost their association with AS for reasons that are as yet still unclear. Other B27 subtypes are very rare and are mostly reported in only single individuals or families, although HLA-B*27:01 and B*27:14 have also been reported in patients with spondylitis. Molecular folding of HLA-B27 protein subtypes is determined by the global effect of polymorphic residues and shows incomplete correspondence to AS.5
HLA-B27 subtype frequencies (%) in different world populations
The association of HLA-B27 with AS is found in nearly all ethnic groups, although it is least strong in those of sub-Saharan African ancestry. Older studies from Africa reported AS to be rare, with a very low frequency of HLA-B27 in patients with AS.6 7 This was attributed, as least in part, to a ‘protective’ West African B27 subtype (B*27:03),7 although subsequent studies reported an association of this subtype with AS in Africans.8 The only previous study of HLA alleles in African Americans with AS suggested that only about 50% of patients are HLA-B27 positive; instead, HLA-B27-negative patients have been shown to be more likely to have the HLA-B7 cross-reactive group antigens (B*7, B*40, B*42, B*55 or B*56).9 Recent studies from West Africa have implicated HLA-B*14:03 in susceptibility to AS.10 This association has also extended to undifferentiated spondyloarthritis and reactive arthritis in the setting of HIV infection where an association is also seen with HLA-B*57:03.10
Several theories have been put forward to explain the association of HLA-B27 and SpA.3 The arthritogenic peptide hypothesis suggests that disease results from the ability of HLA-B27 to bind a unique peptide or a set of antigenic peptides. However, identification of such an ‘arthritogenic peptide’ has been elusive. That said, the current genetic models favour this hypothesis.
Another theory rests on the propensity for HLA-B27 heavy chains to self-associate or form homodimers due to the presence of Cys67 residue (unique to HLA-B27) in their extracellular α1 domain. HLA-B27 heavy chains would then misfold in the endoplasmic reticulum, are retained there by BiP and give rise to a proinflammatory unfolded protein response. Also, HLA-B27 homodimers are expressed on the cell surface and are ligands for a number of natural killer and related cell surface receptors. The fact that most HLA-B27-positive individuals do not develop SpA suggests that this theory is not adequate by itself to explain susceptibility to AS.
A third theory deals with alteration of intracellular invasion/killing of microorganisms. HLA-B27-positive individuals are more efficient at handling certain viral infections (HIV, hepatitis C, influenza) and less able to combat other intracellular bacterial infections (Salmonella, Shigella, Chlamydia, etc). Inability to eliminate these microorganisms contributes to susceptibility to SpA, further suggested by older data demonstrating the presence of bacterial antigens or DNA in the synovium of patients with reactive arthritis.11
Less than 5% of HLA-B27-positive people in the general population develop SpA. On the other hand, 20% of HLA-B27-positive relatives of patients with AS will develop SpA.12 Family studies have suggested that HLA-B27 forms only about 40% of the overall risk for SpA. The entire effect of the MHC, on the other hand, is about 50%. This suggests that other genetic influences are operative in the MHC in addition to HLA-B27.
Other genes implicated in susceptibility to AS
The serological specificity of HLA-B60 (known as HLA-B*40:01 by DNA typing) was shown to be increased in B27-positive patients with AS in five independent data sets in 1989 but not increased in B27-negative patients with AS, increasing the risk for AS threefold (table 2).13 This association was subsequently confirmed in 284 UK patients with AS,14 as well as in HLA-B27-negative Taiwanese patients with AS15 and in African American patients with AS (Zhang et al, unpublished data). However, it was not seen in Mexican Mestizos with SpA.16 Other MHC alleles have also been implicated, but definitive associations with these have been confounded by linkage to HLA-B27.
MHC and non-MHC genes associated with ankylosing spondylitis (updated from Reveille3)
Other than the MHC, a number of genetic influences have been implicated in susceptibility to AS in small cohorts that could not subsequently be replicated. The first gene cluster for which independent confirmation could be established was the interleukin-1 (IL-1) locus on chromosome 2q15. Initial microsatellite data implicated IL-1 receptor antagonist (IL-1RN), although this was not subsequently confirmed. After a larger study instead suggested IL-B, a collaborative study was undertaken of nine single nucleotide polymorphisms (SNPs) in the IL-1 gene cluster, including members IL-1A, IL-1B, IL-1F10 and IL-1RN, in 2675 cases of AS and 2592 healthy controls recruited in 12 different centres in 10 countries, which confirmed that IL-1A is associated with susceptibility to AS (although association of the other IL-1 gene complex members could not be excluded in specific populations).17
More recent studies using computer-automated chip genotyping have allowed denser examination of the genome. A study of British and US patients with AS (the Australo-Anglo-American Spondylitis Consortium or TASC, in collaboration with the Wellcome Trust Case Control Consortium) identified an association with a non-MHC gene encoding an endoplasmic reticulum aminopeptidase, ERAP118 (previously known as ARTS-1). This has been subsequently confirmed in an independent cohort from the UK, as well as in additional patients from the USA, Canada, Portugal, Hungary and China.19,–,23
ERAP1 has two known functions, either of which may explain its association with AS. Within the endoplasmic reticulum, ERAP1 is involved in trimming peptides to the optimal length for MHC class I presentation.24 This would suggest that antigen presentation is crucial in the pathogenesis of AS. The second known function of ERAP1 is that it cleaves cell surface receptors for the proinflammatory cytokines IL-1 (IL-1R2), IL-6 (IL-6Rα) and TNF (TNFR1), thereby downregulating their signalling.25
Based on the observation that the IL-23 receptor (IL-23R) locus was associated with IBD26 and subsequently psoriasis,27 an analysis was performed of seven SNPs in the IL-23R gene in 532 UK and 903 North American patients with AS. The most significant association was with the same SNP found in IBD, rs11209026. This SNP was associated as part of an extended haplotype and not independently. The association of IL-23R with AS has subsequently been confirmed in studies from Canada, Spain, Portugal and Hungary, although it was not associated with AS in Koreans and inconsistently in two studies from China.21 23 28,–,31
In a follow-up study of 2300 British, US and Australian cases of AS of white European descent examining 370 000 SNPs using Illumina HumHap300 microarray genotyping slides, the ERAP1 and IL-23R associations were extended and additional regions were implicated in AS susceptibility, including gene deserts at chromosome 2p15 and 21q22 and additional candidate genes IL-1 receptor 2 (IL-1R2) and anthraxin receptor 2 (ANTXR2)32 (figure 1).

Innate immunity, antigen presentation and the Th17 pathway all implicated in susceptibility to ankylosing spondylitis (AS).
The gene desert at 2p15 has not previously been associated with any other disease and there are no other genes in the immediate vicinity. The gene desert at 21q22 has previously been associated with paediatric IBD,33 although the association in our dataset did not change with exclusion of clinical IBD cases. As with 2p15, there are no genes in the immediate vicinity.
Another gene implicated in this study was IL-1R2, which is not associated with any other disease. It acts as a decoy receptor, interfering with the binding of IL-1 to IL-1R1. It may be acted upon by ERAP1 in cleavage. IL-1R2 is not differentially expressed in AS, and the association may reflect linkage with IL-1R1 located nearby.
The fourth new gene implicated in this genome-wide association study (GWAS) was ANTXR2. The ANTXR2 gene encodes ANTXR2 (capillary morphogenesis protein-2). The protein binds to collagen IV and laminin, suggesting that it may be involved in extracellular matrix adhesion. Mutations of this gene cause juvenile hyaline fibromatosis and infantile systemic hyalinosis.34 It is not known to be associated with any other disease and is not differentially expressed in AS.
Other SNPs that were increased in frequency but did not achieve genome-wide significance in the TASC study included the gene encoding tumour necrosis factor receptor 1 (TNFR1), which was seen in the discovery set but not in the confirmation set, although it was also seen in a subsequent study in Han Chinese patients with AS. TNFR1 is differentially expressed in AS, and TNF overexpression signals via TNFR1 in mice led to spondyloarthritis. Other SNPs implicated included those in the TNF-associated death domain (TRADD) gene, which was found and confirmed in the UK patients but not North American patients, as well as in CARD9 and STAT3. In a subsequent study of 775 Han Chinese patients with AS and 1587 controls from Shanghai and Nanjing, the associations with ERAP1, STAT3, TNFR1 and the gene desert on 2p15 were further confirmed35 and, in another study examining genes implicated in susceptibility to IBD, associations with KIF21B and CDKAL1 were established.36
Genetic susceptibility in AAU
AAU affects approximately 40% of patients with AS over time and can occur in patients with no evidence of SpA. Over 90% of patients with B27-associated AAU have SpA. Although iritis rarely precedes the clinical onset of AS, it is often the first clue to the recognition that low back pain is inflammatory. Half of patients with anterior uveitis have HLA-B27. A study of microsatellite markers in 76 affected sibpairs with AAU found strong linkage to a region on chromosome 9p21–9p24 and, when compared with a companion cohort of AS families without AAU, the linkage at this region was found in association with AAU but not with AS.37 In addition to this, a GWAS from this group has implicated ERAP1 and IL-23R as well as novel genes whose confirmation is pending.
Genetics of psoriasis and PsA
PsA is another disease encountered in the spectrum of SpA and is associated with spondylitis. The prevalence of psoriasis is much higher among first-degree relatives of probands with PsA than in the general population. Moreover, in one recent study, the recurrence risk ratio for first-degree relatives (λ1) for PsA was 30.4 and that for psoriasis was 7.6, suggesting a stronger heritability for PsA than psoriasis per se.38 Early studies implicated HLA-B13, B37 and B57, which subsequently were found to be in linkage disequilibrium with HLA-Cw6. The HLA-Cw6 association is specifically seen in those with plaque and guttate (although not with palmoplantar) psoriasis and indicates higher risk for younger age at onset, familial aggregation and more severe disease. HLA-Cw6 preferentially presented cross-reactive peptides from streptococcal M protein and the hyperproliferative keratin K17 to skin homing CD8 T cells, suggesting that the evolution of guttate into chronic plaque psoriasis might reflect a transition from a self-limited response to Streptococcus initiated in the tonsils to a sustained response to homologous peptides derived from hyperproliferative skin keratins.39
Previous family studies using microsatellite markers implicated the most important region of psoriasis susceptibility mapped to a 60 kb interval telomeric to HLA-C known as PSORS1.40 Sequencing of this region has implicated HLA-C itself.41 Environmental factors such as trauma, stress and infections such as streptococcal pharyngitis have also been implicated. GWASs in psoriasis have implicated a number of genes encoding cytokines and cytokine pathways in psoriasis susceptibility (table 3, figure 2).42 43 A recent GWAS in Caucasians has identified genes in the Th17 pathway including IL-12B (the p40 subunit of IL-23 and IL-12), IL-23A (the p19 subunit of IL-23) and IL-23R (encoding a subunit of the IL-23 receptor), as well as genes in the nuclear factor κB (NFκB) pathway (TNFAIP3-TNF-α induced protein 3 and TNIP1-TNFAIP3 interacting protein 1) and genes in the Th2 pathway (IL-4, IL-13).42 A concomitant study in Chinese patients with psoriasis replicated the MHC and IL-12B associations and identified a new susceptibility locus within the LCE gene cluster on 1q21,43 which contains multiple conserved genes that encode stratum corneum proteins and are expressed relatively late during fetal assembly of the skin cornified envelope. Most recently, the Wellcome Trust Case Control Consortium 2 and the Genetic Analysis of Psoriasis Consortium have identified multiple new loci in psoriasis susceptibility. Independent confirmation will be necessary to better understand their role in psoriasis vulgaris and PsA susceptibility.44
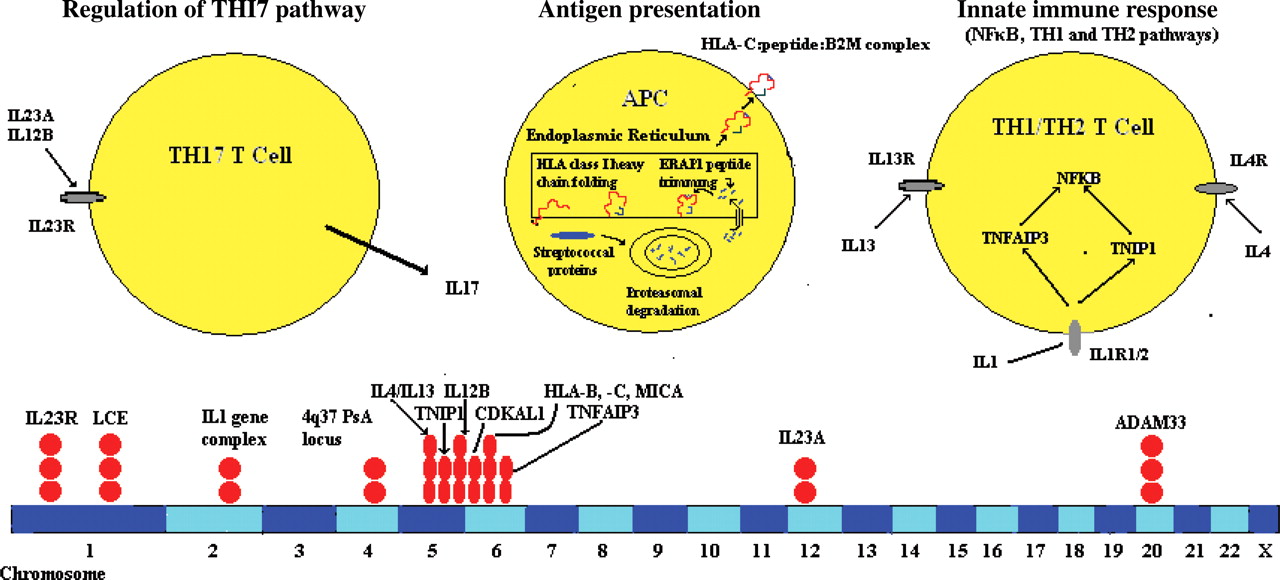
Innate immunity, antigen presentation and the Th17 pathway all implicated in susceptibility to psoriasis and psoriatic arthritis.
MHC and non-MHC genes associated with psoriasis and psoriatic arthritis (updated from Reveille3)
Probably due to phenotypic heterogeneity and case ascertainment, the genetics of PsA is not as well defined, although associations with HLA class I alleles (ie, HLA-B38, B39 and B27) have consistently been reported. Studies from the UK and Canada have implicated genes in the IL-1 gene complex45 46 as well as IL-13,47 48 and a recent GWAS has identified a region at chromosome 4q27 that has been implicated in a number of autoimmune diseases.49
Genes and IBD susceptibility
Sacroiliitis and spondylitis occur in up to 20% of patients with IBD. Moreover, studies from Europe have shown that 22–69% of patients with AS have microscopic evidence of gut inflammation, although only about 7% develop Crohn's disease.50 51
The most consistent region for IBD susceptibility on genome-wide screens has been found on chromosome 16q12 (IBD1) at NOD2/CARD15,52 53 whose product serves as an intracellular receptor for bacterial products in monocytes and transduces signals leading to NFκB activation. Mutant alleles of NOD2 have been associated with susceptibility to Crohn's disease in most ethnic groups; 10–30% of patients with Crohn's disease are heterozygotes and 3–15% are homozygotes for one of three NOD2 mutations compared with 8–15% and 0–1% of controls, respectively. NOD2 explains about 20% of the overall genetic susceptibility.
More recently, genes located in a functional haplotype in the susceptibility region IBD5 on chromosome 5, including the genes prolyl 4-hydroxylase through interferon regulatory factor 1 and including the organic cation transporter (OCTN) gene cluster has been identified and confirmed.54 Although the precise mechanism is not known, mutations in NOD2/CARD15 or OCTNI/II may affect the host's ability to combat bacteria that gain entry to the host through the gut.
GWASs have also implicated genes in the Th17 pathway such as IL-23R on chromosome 1p31, a finding that has been reproduced extensively in replication cohorts of patients with Crohn's disease or ulcerative colitis,26 and others such as STAT3. More recently, an intergenic region on chromosome 10q21.1 and a coding variant in ATG16L1 (2q37), which is expressed in intestinal epithelial cell lines, have been implicated and replicated in IBD susceptibility.55 56 Functional knockdown of this gene abrogates autophagy of Salmonella typhimurium.56 Dendritic cells (DCs) from patients with Crohn's disease with susceptibility variants in NOD2 or ATG16L1 are deficient in autophagy induction, suggesting that NOD2 influences bacterial degradation and interacts with the MHC class II antigen presentation machinery within DCs, and that ATG16L1 and NOD2 are linked within a functional pathway.57 Also reported were strong associations with independent replication to variation in the genomic regions encoding PHOX2B, NCF4 and a predicted gene on 16q24.1 (FAM92B) (figure 3). Most recently, a number of new genes have been identified in Crohn's disease and UC susceptibility by GWAS and meta-analysis approaches, bringing the total of IBD susceptibility loci up to 7158,–,60; their complete delineation is outside the scope of this review. However, only 25% of the genetic variance for IBD is explained by the genes ascertained thus far, suggesting a need for future studies to identify rare variants and recessive traits not easily identifiable by GWAS approaches.

{kind=link}
{kind=link}
{kind=link}
Innate immunity, autophagy and the Th17 pathway all implicated in Crohn's disease.
Conclusions and future directions
Significant advances have been made recently in elucidating the genetic basis of SpA. In AS, along with recent studies confirming other MHC (ie, B*40:01) and non-MHC (IL-1A) influences in addition to HLA-B27, GWASs have shown an important impact of other genes such as ERAP1, IL-23R as well as other genes and genetic regions (gene deserts on chromosomes 2 and 21, IL-1R2, ANTXR2 and STAT3, TNFR1, TRADD, CARD9). In psoriasis and PsA, the MHC component has also been dissected and is likely to include at least HLA-C and possibly other MHC influences. In addition, TH17 cytokine genes in common with AS (IL-23R) and unique to Ps (IL-12, IL-23A), as well as TH2 cytokines (IL-4, IL-13) and NFκB pathway genes (TNFAIP3, TNIP1), have been implicated. In IBD, genes important in the innate immune response (NOD2), autophagy (ATG6L1) and regulation of the IL-23 pathway (IL-23R) play a role in disease susceptibility. These data suggest that genetic markers in susceptibility to AS, psoriasis and IBD are both disease-specific and class-specific, suggesting common mechanisms of susceptibility and pathogenesis. There is also the possibility that these markers could be used along with HLA testing (particularly B27) in diagnosis. However, apart from the MHC genes, the individual contributions of individual non-MHC genes or SNPs is quite small, the causal variants are yet to be identified, and much of the genetic variance is still unexplained. Hence, the use of genetic testing outside HLA-B27 is premature.
References
Footnotes
-
Funding This study was supported by grant P01-052915-01.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.