Article Text

Abstract
Background: It has been suggested that bacterial infections have a role in the pathogenesis of rheumatoid arthritis (RA). P gingivalis, a Gram-negative, anaerobic rod, is one of the major pathogens associated with periodontal disease.
Objective: To examine P gingivalis infection and its effects on cell cycle progression and apoptosis of human articular chondrocytes.
Methods: Primary human chondrocytes cultured in monolayers were challenged with P gingivalis. Infection and invasion of P gingivalis into chondrocytes was analysed by scanning electron microscopy, double immunofluorescence and by antibiotic protection and invasion assay. Cell cycle progression of infected chondrocytes was evaluated by flow cytometry. Also, cell apoptosis was visualised by terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) of DNA strand breaks and by western blot analysis.
Results: Data showed that P gingivalis could adhere and infect primary human chondrocytes. After chondrocyte infection, intracellular localisation of P gingivalis was noted. Flow cytometry analyses demonstrated affected cell cycle progression, with an increase of the G1 phase and a significant decrease of the G2 phase after infection. In addition, increased apoptosis of P gingivalis-infected chondrocytes was visualised by TUNEL assay and by upregulation of caspase-3 protein expression.
Conclusion: These data demonstrate that P gingivalis infects primary human chondrocytes and affects cellular responses, which might contribute to the tissue damage seen in the pathogenesis of rheumatoid arthritis.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) is a systemic inflammatory disorder characterised by accumulation of proinflammatory cell infiltrates in the synovial membrane, leading to synovitis and destruction of cartilage and bone tissue of the joints.1 2 Genetic and environmental factors such as age, gender, smoking as well infections seem to have a role in the pathogenesis of RA.3 Although bacterial infections, in general,3 4 5 6 7 8 and oral pathogens, in particular,9 10 11 12 have been related to the pathogenesis of RA, the pathomechanisms involved are unknown.
Inflammatory periodontal disease (PD) is a chronic bacterial infection of the periodontal tissues mainly caused by Gram-negative, anaerobic bacteria, resulting in soft and hard periodontal tissue destruction.13 PD has recently been linked to RA by our group and others; however, so far evidence of disease association is almost entirely based on epidemiological data.14 15 16 It has been recognised that RA and PD share a number of pathophysiological features such as a common proinflammatory trait—for example, increased synthesis of inflammatory mediators as well as mechanisms of tissue destruction.9 17 18 Also, both diseases have common genetic predispositions such as increased association with HLA-DRB1 polymorphism.18
The oral cavity harbours a large reservoir of pathogenic organisms that may contribute to chronic bacteraemia and potential damage of distant organs, including the joints.4 19 Also, there is evidence of a specific antibody response against anaerobic oral bacteria as well as the presence of oral bacterial DNA in the synovial fluid of patients with RA.10 20
Porphyromonas gingivalis (P gingivalis), a Gram-negative anaerobic black-pigmented rod, is one of the major pathogens associated with PD.21 P gingivalis has been shown to attach and to invade epithelial22 23 and endothelial cells as one possible mechanism to enter the bloodstream and to exert systemic effects.24 Also, P gingivalis has been shown to induce serum rheumatoid factor12 and it seems to be the only pathogen known that expresses peptidyl arginine deiminase, a central susceptibility factor for RA.25 Limited data suggest that P gingivalis induces chondrocyte proteoglycan degradation.26 27 The aim of this study was to investigate direct cellular effects of P gingivalis on chondrocyte cell cycle progression and apoptosis, as a potential pathomechanism contributing to joint damage seen in RA.
Materials and methods
Chondrocyte isolation and culture
Chondrocyte isolation was performed as described previously.28 29 Cartilage was obtained from knee joints of donors with degenerative joint disease. Experimental protocols were approved by the ethics committee of the Charité-Universitätsmedizin, Berlin, Germany. Cartilage was minced and digested in medium containing 1 mg/ml clostridial collagenase (Sigma-Aldrich, Deisenhofen, Germany) and 1 mg/ml pronase (Sigma-Aldrich) for 30 min at 37°C each. Digested solution was then filtered, centrifuged and the cell pellet was suspended in Hams F12/10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and cultured to subconfluence (80%) at 37°C, 95% air and 5% CO2 before use.
Bacterial cultivation
P gingivalis ATCC 33277 (Manassas, Virgina, USA), tested by Hain Life Science (Nehren, Germany) and by a microbiological test kit ID 32A (BioMérieux, Marcy l’Etoile, France), was cultivated as described before.24 Stocks were grown in brain heart infusion broth containing 0.1% cysteine, 0.5% yeast, 0.1% vitamin K and 0.25% haemin in an anaerobic chamber (Thermo Life Sciences, Lund, Sweden) in an atmosphere of 85% N2, 10% CO2 and 5% H2. Aliquots of P gingivalis were grown to mid-logarithmic phase, centrifuged and resuspended in Dulbecco’s modified Eagle’s medium HAMS F12 medium. A multiplicity of infection of 100 was used.
Scanning electron microscopy
Infected chondrocytes were grown on glass slides and fixed in Karnowsky’s solution (2% glutaraldehyde, 4% paraformaldehyde in phosphate-buffered saline (PBS)) at 4°C. After washing with HEPES buffer, samples were dehydrated with graded ethanol, critical-point dried with CO2, sputter coated with 10 nm of gold and examined with a scanning electron microscope with 5 kV in the secondary mode (LEO 1530 Field emission scanning electron microscopy; LEO Elektronenmikroskopie GmbH, Oberkochen, Germany).
Intracellular detection of P gingivalis by double immunofluorescence
Double immunofluorescence was performed, as described previously.30 31 Infected chondrocytes were grown on glass coverslips and fixed with 3% paraformaldehyde (pH 7.6) for 20 min. Without prior permeabilisation, samples were blocked and incubated with 1:200 primary antibody against P gingivalis for 30 min. Primary antibody specific for P gingivalis was a generous gift from Professor C Genco (Department of Microbiology, Boston University, Massachusetts, USA). Samples were then treated with green fluorescent secondary antibody A 488 for 12 h at 4°C. Subsequently, samples were permeabilised with 1% Triton X100 for 15 min to allow intracellular staining of P gingivalis. After permeabilisation, cells were incubated with 1:200 primary antibody against P gingivalis for 30 min and then treated with red fluorescence secondary antibody A 546 for 12 h. Slides were mounted with Permafluor (Beckman-Coulter, Fullerton, California, USA) and observed by Zeiss LSM 5 Pascal, Objective: 63× NA:1.4 Plan-Apochromat (Carl Zeiss, Jena, Germany). Cell images were captured and image analysis was performed by LSM 5 version 2.8 imaging software (Zeiss).
Antibiotic protection and invasion assays
Invasion of chondrocytes by P gingivalis was quantified by determining the number of colony forming units (CFUs) recovered after antibiotic treatment as described.24 32 Chondrocytes were infected with P gingivalis for 2 h, non-attached bacteria were removed by washing and half of the monolayers were lysed with sterile H2O. Lysates were diluted and plated on Columbia agar plates containing 0.1% vitamin K and 0.25% haemin and incubated for 7 days under anaerobic conditions. On the other half of the monolayers, external adherent bacteria were killed by 0.1 mg/ml metronidazole and 0.5 mg/ml gentamicin. After 4 h, the remaining chondrocytes were also lysed, plated on agar plates and CFUs of invasive bacteria were then counted. Invasion was expressed as the percentage of the initial inoculum recovered after antibiotic treatment and chondrocyte cell lysis.
Cell cycle analysis
For cell cycle analysis, infected chondrocytes were trypsinised, washed with PBS containing 2% FBS, and fixed in 100% ethanol for 20 min at 4°C. Cells were stained with propidium iodide (50 μg/ml) in PBS containing 2% FBS and 0.5 mg/ml RNase A and were analysed by flow cytometry using a FACScan flow cytometry with CELLQUEST acquisition and analysis software (BD Biosciences, San Jose, California, USA).
In situ cell death detection assay
A terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) technique (Roche, Mannheim, Germany) was used to detect chondrocyte cell apoptosis.30 33 Chondrocytes, grown on 24-well plates at a density of 2×104 per well, were infected with P gingivalis. Staurosporine (1 μM), as inducer of cell apoptosis, was used as positive control. Infected cells were fixed, permeabilised with 0.1% Triton X100 in Na citrate (0.1 M; pH 6.0) and TUNEL reaction mixture was added for 60 min. For stabilisation of FITC-fluorescence, cells were treated with Alexa 488-labelled anti-fluorescein antibody for 60 min at 37°. F-actin was visualised by phalloidin A 546 (Molecular Probes, Eugene, USA) staining. Slides were mounted, observed by Zeiss LSM and images were analysed by LSM 5 version 2.8 imaging software (Zeiss).
Western blot analysis
Infected chondrocytes were harvested with cold lysis buffer comprising 1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 0.25 mM EDTA, 1 mM phenylmethylsulphonyl fluoride, 10 μM each of antipain, leupeptin and pepstatin A. Protein concentration was determined by the Bradford method (Bio-Rad Laboratories, Hercules, USA). Protein samples were resuspended in gel-loading Laemmli buffer (Bio-Rad Laboratories), protein extracts (80 μg/lane) were fractionated by 10% sodium dodecyl sulphate-polyacrylamide electrophoresis gel (SDS-PAGE) and blotted onto Hybond-ECL membranes (Amersham, Dreieich, Germany). The membranes were blocked with Odyssey blocking buffer (Li-Cor, Lincoln, USA), then incubated overnight with blocking buffer containing rabbit polyclonal caspase-3 antibody (Santa Cruz Biotechnologies, California, USA) and mouse monoclonal ERK2 antibody (Santa Cruz) as loading control, each at a dilution of 1:2000. The next day, the membranes were incubated for 1 h with blocking buffer containing fluorochrome-conjugated secondary antibodies: CY5.5-conjugated anti-mouse IgG and IRDye 800-conjugated anti-rabbit IgG (Rockland Immunochemicals, Pennsylvania, USA), each at a dilution of 1:2000. Proteins were visualised by using an Odyssey infrared imaging system (Li-Cor). Protein sizes (caspase-3 20–22 kDa, ERK2 43–44 kDa) were confirmed by comparison with a protein molecular weight marker (Bio-Rad’s SDS-PAGE Standard, Bio-Rad Laboratories).
Release of lactate dehydrogenase (LDH)
Chondrocyte monolayers were challenged with P gingivalis, 1 μM staurosporine (negative control) and 2% Triton X 100 (positive control). LDH release in the supernatant was determined by the colorimetric measurement of the reduction of sodium pyruvate in the presence of NADH and expressed as the percentage of total enzyme activity liberated from chondrocytes in the presence of P gingivalis.
Statistical analysis
An unpaired Student t test assuming equal variances was used as indicated in the legends. A p value of <0.05 was considered significant.
Results
P gingivalis infection of human chondrocytes
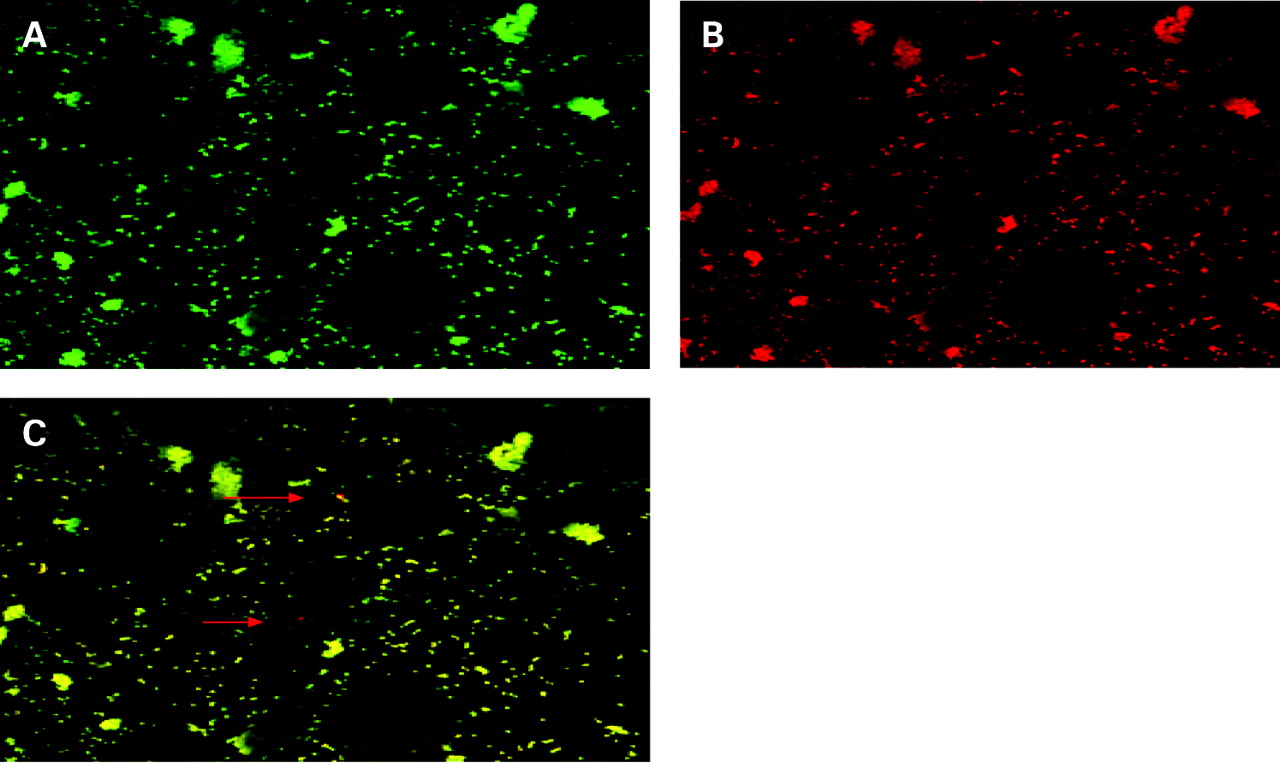
To visualise infection and adhesion of P gingivalis to primary human chondrocytes, scanning electron microscopic analysis was performed. Two hours after infection, close adherence of P gingivalis to the surface of primary human chondrocytes was found (fig 1). In addition, by double immunofluorescence and LSM analysis, single rods of intracellular P gingivalis were observed at 2 h after infection (fig 2). To further quantify invasion and infection of chondrocytes by P gingivalis, a modified antibiotic protection and invasion assay was applied, as described in “Materials and methods”. P gingivalis (7.7×104 CFU/ml) could be recovered within the chondrocyte cell lysates (intracellular bacteria), representing an invasion rate of 27.5% relative to the total amount of bacteria added (5×107 bacteria/ml) (fig 3).

Infection of primary human chondrocytes by P gingivalis demonstrated by scanning electron microscopy. Chondrocytes were infected for 2 h with a multiplicity of infection of 100 as described in “Material and methods”. Note adherence and interaction of P gingivalis with chondrocytes. Experiments were performed at least three times. A representative image (A) and a closer view (B) of one experiment are shown.
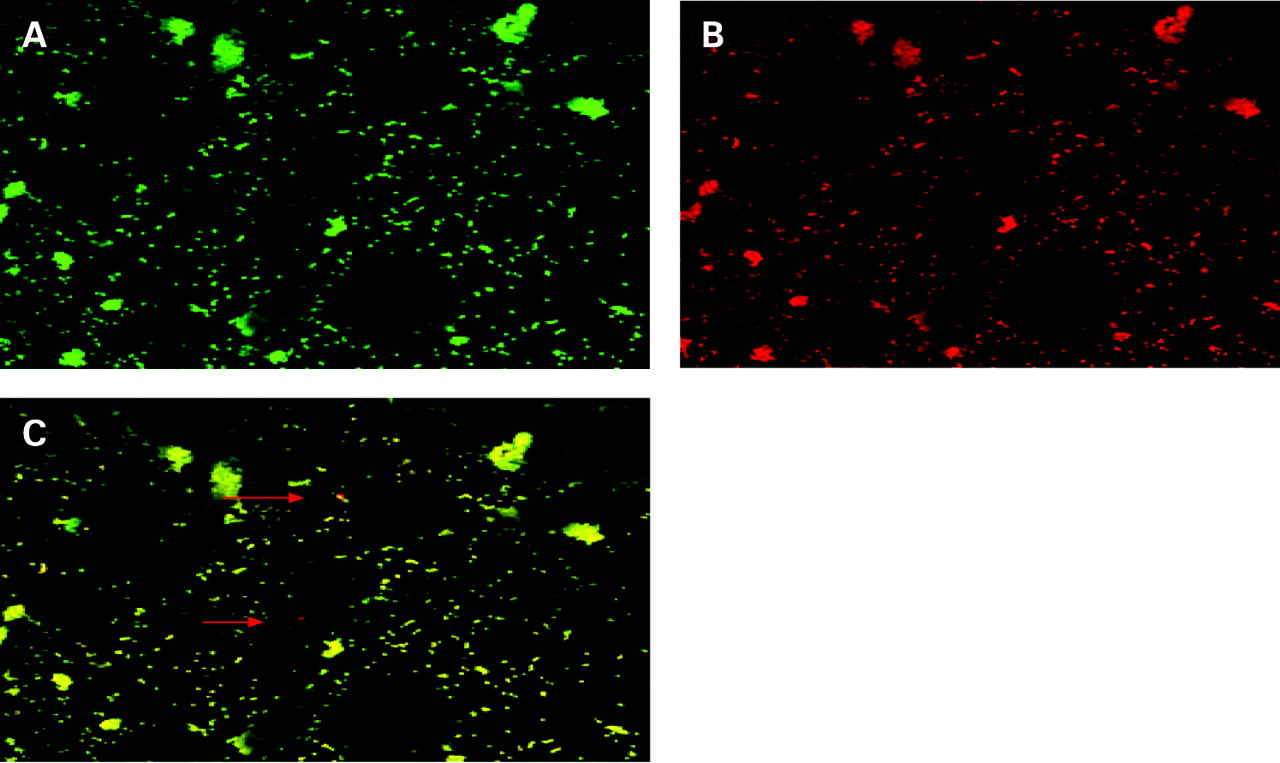
Intracellular P gingivalis visualised by double immunofluorescence. Primary human chondrocytes were infected with P gingivalis for 2 h. After fixation, extracellular P gingivalis were labelled (green fluorescence) (A), subsequently chondrocytes were permeabilised and extracellular and intracellular P gingivalis were labelled (red fluorescence) (B). (C) Double immunofluorescence which demonstrates the presence of intracellular P gingivalis (red fluorescence, red arrows). Experiments were performed at least three times. A representative image of one experiment is shown.

Invasion of P gingivalis analysed by an antibiotic protection assay. P gingivalis was added to human chondrocytes (multiplicity of infection = 100) and further incubated as described in “Materials and methods”. The total represents bacteria that adhered or already invaded after 2 h of exposure to chondrocytes. EC indicate those bacteria which only adhered to chondrocytes. IC indicates bacteria found in the chondrocyte cell lysates after 2 h of incubation followed by antibiotic treatment for 2 h. Colony forming units (CFUs; 7.7×104/ml) of intracellular P gingivalis could be recovered from the chondrocyte cell lysates. Data are presented as mean (SD) of three separate experiments.
P gingivalis affects chondrocyte cellular responses
As P gingivalis adhered and invaded human chondrocytes, we further investigated its effects on cellular processes. Figure 4 shows an increase of the percentage of cells in the G1 phase as well as a decrease of cells in the S phase and a significant (p = 0.054) decrease of the G2M phase after infection compared with control cells.

Effects of P gingivalis on cell cycle progression. After P gingivalis infection at an multiplicity of infection of 100 and subsequent antibiotic treatment, cell lysates were subjected to flow cytometric analysis after 24 h. (A) Representative diagram of control cells. (B) Representative diagram of P gingivalis-infected cells. An increase of the percentage of cells in the G1 phase and a decrease of cells in the S as well as in the G2 phase of the cell cycle was noted after infection. Data are presented in per cent as mean (SD) of three separate experiments. *p = 0.054 versus control (G2/M phase) by an unpaired Student t test assuming equal variances.
In addition, cell apoptosis after P gingivalis infection was studied. TUNEL analysis showed that P gingivalis induced DNA fragmentation 2 h and 4 h (fig 5) after infection compared with non-infected chondrocytes. In addition, cytoskeletal rearrangements and increased stress fibre formation was noted beginning 2 h after P gingivalis infection (fig 5). In addition, compared with control cells, increased expression of caspase-3 protein, one of the central executioner caspases, was found at 1 h and 2 h after P gingivalis infection (fig 6).

Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) stain demonstrates apoptosis (green fluorescence) in P gingivalis infected human chondrocytes. Chondrocyte monolayers were cultivated in the absence (A) or presence of P gingivalis for 2 h and 4 h (C, D). Apoptosis was observed at 2 h (C) and at 4 h (D) after infection. Staurosporine (1 μM), as positive control, induced apoptosis (B) F-actin, visualised by phalloidin (red fluorescence), demonstrates stress fibre formation after infection. Experiments were performed at least three times. A representative image of one experiment is shown.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Caspase-3 protein expression after P gingivalis infection. Chondrocytes were infected with P gingivalis for 30 min, 1 h and 2 h, and cell lysates were subjected to western blot analysis. Staurosporine (1 μM) served as positive control. Induced caspase-3 expression was found 1–2 h after infection. Simultaneous detection of ERK2 documented the protein load. Experiments were performed at least three times. A blot of one representative experiment is shown.
Also, to rule out non-specific DNA fragmentation in late cell apoptosis, we analysed LDH release in the cell supernatant. Compared with control (0.87 (0.05)), we noted insignificant LDH release upon 2 h of P gingivalis infection (0.89 (0.03)) as well as after exposure to staurosporine (0.91 (0.04)). At 4 h a significant (p<0.05) increase of LDH release was measured (1.6 (0.09)), indicating the start of cell necrosis.
Discussion
Bacteraemia with oral pathogens occurs frequently in patients with inflammatory PD34 and may contribute to increased risk of distant organ infections, including inflammatory joint diseases.9 Oral infections have been related to the pathogenesis of RA.9 10 11 12 It has been speculated that infectious agents such as P gingivalis in association with a genetic predisposition might serve as a direct trigger of joint tissue destruction seen in RA or indirectly modify immune responses after disease onset, or a combination of both.9 35 Oral bacterial DNA has been detected in the synovial fluid of patients with RA,10 which may indicate that penetration to the joint cavity to promote direct bacterial effects on articular cells, including chondrocytes, is a potential pathway in the pathogenesis of RA.
These data show for the first time that P gingivalis can adhere to and infect primary human chondrocytes in an experimental setting. Also, intracellular bacteria of P gingivalis were found. Adhesion and invasion have been shown to be common strategies of molecular interaction between P gingivalis and a variety of host cells, including endothelial, and epithelial cells, macrophages and fibroblasts.24 36 37 38 In this study, an invasion rate of 27.5% into human chondrocytes was noted. In other mesenchymal cells of human origin, invasion rates of between 2.9% and 34% have been reported for P gingivalis.24 36 37 Invasion rates vary depending on the P gingivalis strain.36 In this study, P gingivalis strain 33277 was applied, which has been frequently used in invasion studies24 36 37 and which has been shown to be highly invasive in mesenchymal cell types.36 In addition, invasion mechanisms seem to vary slightly between different cell types. In mesenchymal cells, P gingivalis is internalised in phagosomes and later sequestered,39 while in epithelial cells internalised cells are found freely in the cytoplasm or within single membrane-bound vacuoles.23 40 Present results of double immunofluorescence and LSM imaging indicate a rather solitary location of P gingivalis within the cytoplasm of human chondrocytes. In this study, freshly isolated human chondrocytes were used for all experiments to assure reproducible infections. The approach with primary cells, compared with established cell lines, seems to have beneficial effects on the invasion efficiency by P gingivalis.23 40 41 Also, it is important to note that in this study intracellular P gingivalis could be recultivated after chondrocyte cell lyses, which indicates that internalised P gingivalis survives and stays viable within the chondrocytes for the time studied, as shown for other cell types.24 32 36 40 Inflammatory tissue destruction in the rheumatoid joint is a result of an interaction of different cells types.42 43 Chondrocytes have a central role in cartilage matrix destruction as they respond to proinflammatory cytokines released from the synovium as well as producing mediators that promote tissue catabolism.42 Also, it would be of interest to determine whether other articular cells including synovial fibroblasts43 are affected by P gingivalis.
The intracellular location of bacterial pathogens suggests that they have an impact on cellular processes. These data demonstrate that P gingivalis affects cell cycle progression of human chondrocytes, producing a trend towards an increase of the percentage of cells in the G1 phase and a decrease of cells in the S and G2M phases. Recently, studies in other cell types showed that P gingivalis can manipulate the host cell cycle.44 45 In osteoblasts, P gingivalis inhibited cell cycle progression by arresting cells in the G1 phase. The G1 phase is important for signals that regulate both the exit from the cell division cycle to differentiation and the reactivation of cell proliferation.46 The progression of the cell cycle phases is precisely regulated to coordinate normal cell division.47 A delay of the cell cycle progression by P gingivalis could have significant effects on the integrity and turnover of the articular cartilage tissue, favouring tissue degradation.
In this study, increased apoptosis of P gingivalis-infected primary human chondrocytes was demonstrated by morphological and by biochemical analysis, including DNA fragmentation and activation of effector caspase-3. A number of intracellular pathogens, including oral bacteria such as P gingivalis, have been shown to regulate apoptosis and impinge upon a number of tightly regulated apoptotic pathways—including caspases—of infected mammalian cells.37 48 49 50 51 Intracellularly, one major apoptotic pathway is the regulation of a family of cysteine proteases (caspases).52 These results demonstrate that caspase-3 is increased 1–2 h after infection. Accordingly, maximal DNA fragmentation, accompanied by insignificant LDH release, was seen 2 h after P gingivalis infection, which indicates apoptosis and the absence of cell necrosis. In agreement with our results, P gingivalis induced apoptosis of gingival epithelial cells 2 h after infection.49
In addition, it is important to note that P gingivalis produces a variety of different virulence factors.53 Whether whole bacteria, specific virulence factors or a combination of both primarily interact with chondrocytes remains to be determined.
In conclusion, this experimental study demonstrates that P gingivalis has the potential to infect human chondrocytes. Whether affected cell cycle progression and apoptosis by chronic bacterial infections may have a role in the initiation or progression of inflammatory joint disease, and therefore possibly contribute to the pathogenesis of RA, warrants further studies.
Acknowledgments
Some of this research were conducted by ER in partial fulfilment of the requirements for an MD degree from Charité-Universitätsmedizin Berlin, Germany.
REFERENCES
Footnotes
NP and ER contributed equally to this work.
Funding This work was financially supported by DFG GK 325, Bonn, Germany, by DGP e.V., Germany and the support of GABA GmbH, Lörrach, Germany and by a Habilitation Fellowship from Charité-Universitätsmedizin Berlin, Germany to NP.
Competing interests None.
Ethics approval Approval from the ethics committee of the Universitaetsmedizin Charité, Berlin, Germany.