Article Text

Abstract
Background The overall incidence of cancer in patients with rheumatoid arthritis (RA) is modestly elevated. The extent to which cancer rates in RA vary across clinical cohorts and patient subsets, as defined by disease activity or treatment is less known but critical for understanding the safety of existing and new antirheumatic therapies. We investigated comparability of, and means to harmonise, malignancy rates in five RA registries from four continents.
Methods Participating RA registries were Consortium of Rheumatology Researchers of North America (CORRONA) (USA), Swedish Rheumatology Quality of Care Register (SRR) (Sweden), Norfolk Arthritis Register (NOAR) (UK), CORRONA International (several countries) and Institute of Rheumatology, Rheumatoid Arthritis (IORRA) (Japan). Within each registry, we analysed a main cohort of all patients with RA from January 2000 to last available data, and sensitivity analyses of sub-cohorts defined by disease activity, treatment change, prior comorbidities and restricted by calendar time or follow-up, respectively. Malignancy rates with 95% CIs were estimated, and standardised for age and sex, based on the distributions from a typical RA clinical trial programme population (fostamatinib).
Results There was a high consistency in rates for overall malignancy excluding non-melanoma skin cancer (NMSC), for malignant lymphomas, but not for all skin cancers, across registries, in particular following age/sex standardisation. Standardised rates of overall malignancy excluding NMSC varied from 0.56 to 0.87 per 100 person-years. Within each registry, rates were generally consistent across sensitivity analyses, which differed little from the main analysis.
Conclusion In real-world RA populations, rates of both overall malignancy and of lymphomas are consistent.
- Rheumatoid Arthritis
- Epidemiology
- Outcomes research
Statistics from Altmetric.com
Introduction
The overall incidence of cancer in patients with rheumatoid arthritis (RA) is modestly elevated, though risks vary by cancer type.1 However, the extent to which cancer rates in RA populations vary across patient subsets as defined by disease activity or treatment is less known but highly relevant for the safety evaluation of new and existing drugs for RA, which require a careful assessment of their risk:benefit ratios. Malignancy represents a critical part of the safety profiles of immunomodulatory drugs. However, typical drug development programmes, with limited sample size and follow-up, offer limited opportunity to assess the risk of low-frequency, long-term outcomes such as malignancies. For ethical reasons, the placebo-controlled period of Phase III studies in RA is typically 6 months or shorter.2 Further, patients in any study arm who do not show a response might be rescued to active treatment. Together, this precludes inferences on long-term safety, including malignancies. Published data from observational/clinical cohorts can be used to provide background rates, but their utility as a comparison or context for trial data is limited by differences in study design, including differences in demographic (eg, age and sex) and geographical characteristics, uncertain comparability with respect to clinical characteristics, suboptimal or uncertain alignment of cancer outcome definitions and the availability of aggregate level (rather than age/sex-specific or patient-level) data only.
The overall aim of this study, which formed part of a larger research programme aimed at improving the means and methods to contextualise drug safety in a typical RA drug development programme, was therefore to investigate the cancer incidence in patients with RA in clinical practice. Specifically, we sought to assess rates of cancer in observational data standardised to the age/sex distribution in a RA trial programme (the Syk inhibitor fostamatinib Phase II/III trial population3 ,4), to investigate the variability of these rates across clinical registries and to investigate the robustness of these rates across defined subpopulations.
Subjects and methods
We (1) identified relevant observational RA cohorts that included individual-level patient data on cancer incidence, (2) harmonised the cancer outcome definitions across these cohorts, (3) identified baseline differences of importance between the cohorts and the clinical trial programme, and the most important predictors for cancer incidence in the cohorts, (4) assembled a matrix of cancer incidence rates stratified by these predictors, enabling the calculation of standardised rates and (5) assessed their robustness across sensitivity analyses including different definitions of sub-cohorts and follow-up.
Study populations
The overall safety contextualisation programme, described elsewhere, linked to the RA trial programme in question (fostamatinib) spanned a range of outcomes.5 Fostamatinib was being developed by AstraZeneca for RA, but discontinued in this indication after its Phase III programme. Since the focus of the current study is on the variability and means to harmonise cancer incidence data from clinical practice rather than on the safety of fostamatinib, we will here refer to it as the ‘RA trial programme’. For contextualisation, five RA registries, described in online supplementary table S1, were selected: Consortium of Rheumatology Researchers of North America (CORRONA) (USA6), Swedish Rheumatology Quality of Care Register (SRR) (Sweden7), Norfolk Arthritis Register (NOAR) (UK8), Institute of Rheumatology, Rheumatoid Arthritis (IORRA) (Japan9) and CORRONA International, based on the following considerations: (1) existing registries/cohorts with a publication track record (except CORRONA International, set-up for this project), (2) global representation to match the geographical profile of a global clinical study programme, (3) size and data quality, including availability of detailed RA-specific longitudinal data, (4) capture of longitudinal data on morbidity and mortality and (5) ongoing data collection reasonably concurrent with the RA trial programme.
Definitions of the study cohort and sub-cohorts
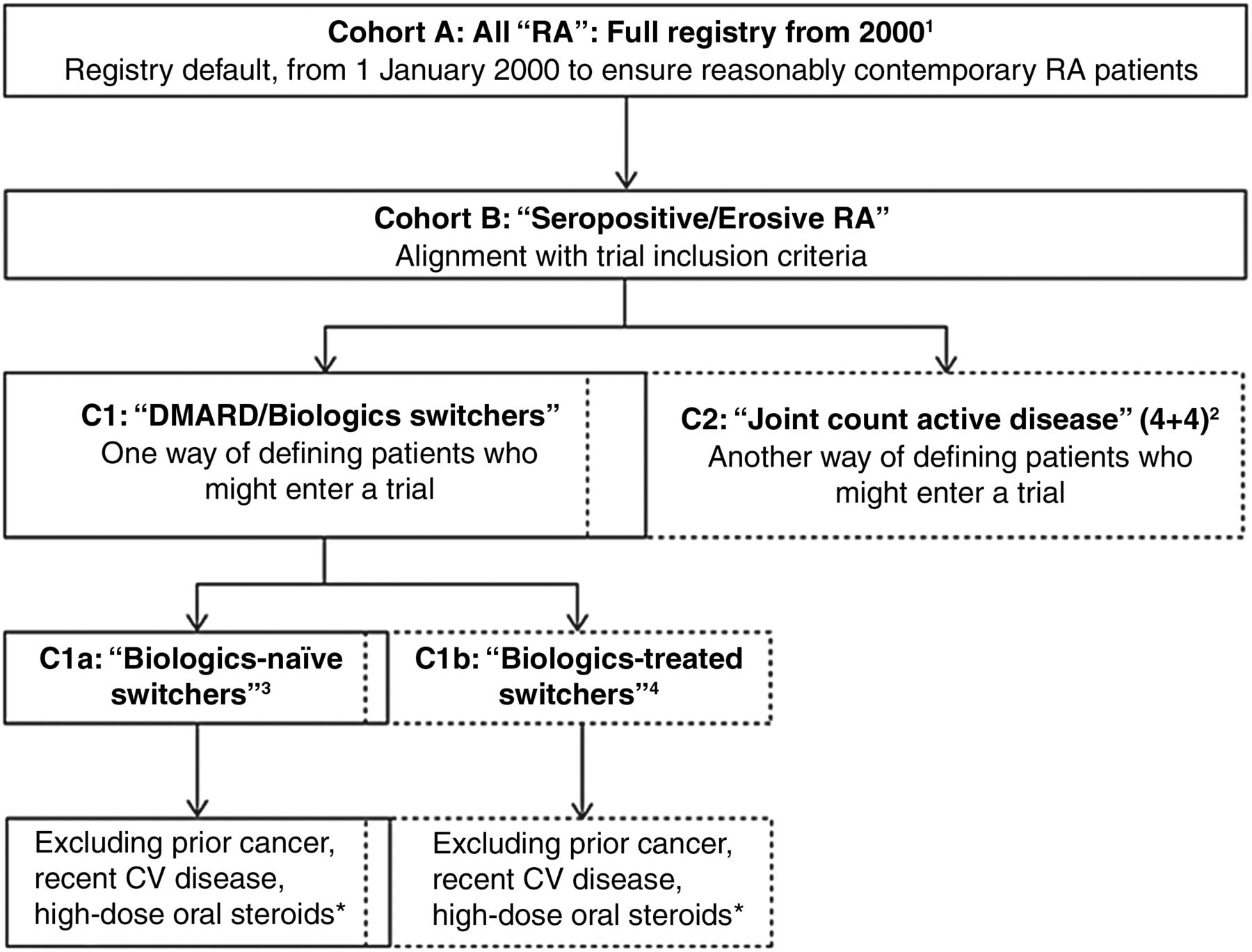
Within each registry, we assembled a main cohort of all patients with RA aged over 18 years who were alive on 1 January 2000 or who joined the registry thereafter but before 31 July 2013. To explore how additional criteria might increase comparability with the RA trial programme, we identified a series of nested sub-cohorts defined by disease activity, treatment status/change and prior comorbidities. Figure 1 illustrates the identification of the main registry cohorts and nested sub-cohorts. No additional inclusion/exclusion criteria were applied.

Definitions and relationship between the main cohort (here denoted Cohort A) and nested sub-cohorts. Note that Cohorts B and C1 are intermediate steps and were not included in the final sub-cohort sensitivity analyses. Note also that the C1 and C2, as well as the C1a and C1b, sub-cohorts are partly overlapping, as some individuals may be included in one sub-cohort at one point in time and then at another (later) point in the other (given the longitudinal character of the data). 1Main registry cohort of patients in registry from 1 January 2000 (or earliest first date after 1 January 2000); baseline at 1 January 2000 or cohort entry. 2Sub-cohort defined by selection of patients with active rheumatoid arthritis (RA) (patients with seropositive/erosive RA and with ≥4 + 4 tender/swollen joints from 28-joint counts), with redefinition of cohort baseline. 3Sub-cohort defined by selection of patients with treatment switch/addition who are previously biologic-naïve (inadequate response to MTX/DMARDs), with redefinition of cohort baseline. 4Sub-cohort defined by selection of patients with treatment switch/addition who have been treated previously with biologics (inadequate response to biologics), with redefinition of cohort baseline. *At time of treatment change/initiation (‘switch’), patients with history of cancer; major CV event in the previous 6 months (myocardial infarction, unstable angina, stroke, pulmonary embolism or heart failure); on oral steroids prednisolone (or equivalent) >10 mg/day—as markers of potential risk for cancer, CV disease and infections were excluded—in alignment with the corresponding relevant trial exclusion criteria. CV, cardiovascular; DMARD, disease-modifying antirheumatic drug; MTX, methotrexate.
Follow-up and cancer outcomes
Start of follow-up was defined as the later of the date on which the patients entered the registry or 1 January 2000. In the nested sub-cohorts, start of follow-up was defined as the later of the date of eligibility for the sub-cohort in question or 1 January 2000. In all analyses, end of follow-up was defined as the earliest of first occurrence of the outcome, loss to follow-up, death, last date of available follow-up data from each registry or 31 July 2013.
We investigated four cancer-related outcomes: all malignancies excluding non-melanoma skin cancer (NMSC), solid malignancies, all skin cancers and malignant lymphomas. Prior to the analysis, event definitions were agreed on for best validity and comparability across registries and the RA trial programme, depending on the type and level of data available (see online supplementary table S2). For benchmarking, we also included cancer rates previously published from the same registries.
Predictors of cancer occurrence in each data source
For each outcome and registry, a series of Cox-regression analyses was performed to gauge the strength of the association between predefined covariates and the outcome under study. The covariates assessed included age, sex, health assessment questionnaire (HAQ) score, body mass index, RA treatment history, indices of RA disease activity at baseline and a set of typical trial exclusion criteria.3 ,4 These analyses indicated which covariates would be the most important to use in the standardisation of incidences from the RA registries. Apart from age and sex, there was no consistent pattern across registries of substantial associations between any of the investigated baseline covariates and the incidence of any of the malignancy outcomes under study (data not shown). Therefore, age (four categories) and sex were used as standardising factors (ie, eight strata).
Assessment of the robustness of the rates
Sensitivity analyses were variations on the main analysis and were performed for the two selected major outcomes of interest: all malignancies excluding NMSC and malignant lymphomas. These analyses applied sub-cohort definitions including selected study inclusion and exclusion criteria to mirror typical criteria in the trial programme (as described above, figure 1), restricted the follow-up time (in calendar time or by truncation at 18 months, see online supplementary table S3) and introduced additional standardisation for HAQ score in three categories (in addition to age/sex; total 24 categories).
Data analysis and estimation of rates
Each registry calculated the number of observed cases of the outcome and the total person-time of follow-up in each stratum defined by the selected standardisation variables. A central analysis used these stratum-specific data to construct incidence rates for each registry, standardised to the distribution of the RA trial programme patient population for the selected standardisation variables, with CIs based on a gamma distribution.10 ,11 All patient-level data remained with the registries throughout.
Results
Baseline characteristics and comparability between the registry cohorts and the RA trial programme population
Table 1 summarises the numbers of patients and available person-time in the main cohorts, each of the nested sub-cohorts and the sensitivity analyses. The main cohorts included between 1564 and 24 176 patients. In each registry, patient numbers dropped considerably (typically by >50%) as additional requirements (eg, about treatment changes or disease activity) were added in sensitivity analyses.
Number of patients (person-years) in the registry cohorts: in the main cohort, in the nested sub-cohorts and remaining sensitivity analyses
Table 2 and online supplementary table S5 summarise the baseline characteristics of patients in the five main cohorts. Across these cohorts, there were marked differences in the age/sex distribution, RA disease duration, disease activity indices, treatment exposures, comorbidities and in the proportions of patients who fulfilled the main exclusion criteria of the RA trial programme. These differences were larger across the registry cohorts than between them and the RA trial programme (see online supplementary table S4). Patients from the RA trial programme were younger, more often women, had higher DAS28 scores, higher HAQ scores, higher swollen joint counts and higher erythrocyte sedimentation rate values than the registry cohorts; the differences in C reactive protein levels were less pronounced (see online supplementary table S4). As in most other RA trial programmes, the inclusion criteria were intended to select subjects with active disease, plus seropositive or erosive disease. In the registry main cohorts, no such restrictions applied.
Demographic and clinical characteristics in five RA registries at baseline (main analysis: main cohort from 1 January 2000). N (%) unless otherwise indicated*
Occurrence, crude and standardised cancer incidence rates across the registry cohorts
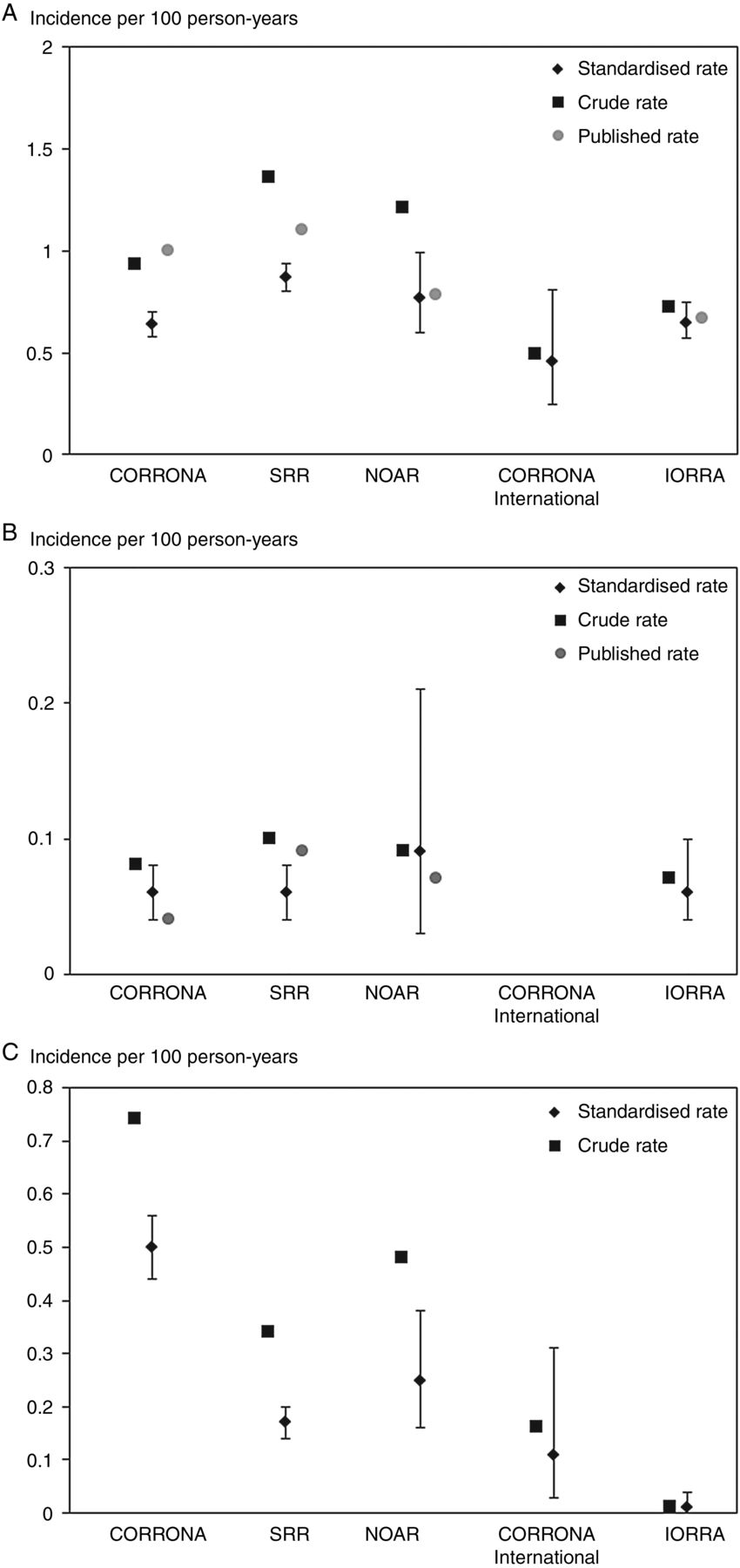
Table 3 summarises the observed numbers of events and the crude and standardised incidences of malignancy across the main registry cohorts (for completeness, data from the RA trial programme are presented in online supplementary table S4). The number of all malignancies excluding NMSC ranged from 129 (NOAR) to 1078 (SRR), in part reflective of cohort size, in part of their age/sex characteristics: the crude incidences for all malignancies excluding NMSC varied approximately threefold, from 0.49 (CORRONA International) to 1.36 (SRR) per 100 person-years (pyr). By contrast, the age/sex-standardised rates varied less (close to twofold). Excluding CORRONA International, the most heterogeneous registry with the shortest follow-up and lowest rate, the age/sex-standardised rates for all malignancies excluding NMSC ranged from 0.64 to 0.87 per 100 pyr (varying by a factor of 1.4).
Numbers of events (person-years), crude and age/sex-standardised incidence rate of malignancy per 100 PY in the registry main cohorts
Age/sex-standardised rates for malignant lymphoma were surprisingly similar, 0.06 per 100 pyr except in NOAR where it was 0.09 per 100 pyr (CORRONA International was not analysed due to insufficient number of lymphoma events). For solid malignancies, age/sex-standardised rates were also remarkably similar. For all skin cancers, however, the age/sex standardised rates varied from 0.01 (IORRA) to 0.50 (CORRONA).
Further sensitivity analyses were restricted to the two major outcomes of interest (all malignancies excluding NMSC and malignant lymphomas). CORRONA International provided too few events and was excluded from subsequent analyses.
Robustness of the standardised incidence rates
Table 4 shows the age- and sex-standardised cancer incidence rates across sensitivity analyses (including HAQ standardisation in one sensitivity analysis) and across registries for all malignancies excluding NMSC and for malignant lymphomas. For all malignancies excluding NMSC, rates varied somewhat across the sensitivity analyses, from 0.59 to 0.84 (CORRONA), 0.73 to 1.14 (SRR), 0.46 to 0.77 (NOAR), 0.46 to 0.66 (CORRONA International) and 0.51 to 0.71 (IORRA) per 100 pyrs. Across registry cohorts, there was no obvious pattern of highest/lowest rates observed in any of the sensitivity analyses, with the possible exception of higher rates (CORRONA and SRR) in the sensitivity analyses restricted to previously biologics-treated patients who added/switched treatment (table 4).
Summary of all malignancies excluding non-melanoma skin cancer, and of malignant lymphomas; incidence rates per 100 person-years standardised by age and sex across all sensitivity analyses
For malignant lymphomas, the number of events was smaller (table 3), the CIs were wider and the variation across registries/sensitivity analyses somewhat more difficult to interpret. However, there was no consistent pattern of variation except higher point estimates in the sensitivity analyses restricted to sub-cohorts of previously biologics-treated patients who added/switched treatment (table 4).
Previously published rates from the registries included in this study
While several previous publications had described relative risks, cancer incidences in the different RA cohorts themselves or in subsets thereof were less often provided, and if so, typically as overall rather than age/sex-specific rates. Figure 2A–C illustrates the relationship between the crude incidence rates (reflective of the age/sex composition of each registry); the age/sex-adjusted incidence rates obtained in our study (ie, standardised to the RA trial programme) and the previously published (crude) rates from the same registries.12–19 The difference between these three estimates reflects the level of ‘harmonisation’ achieved via age/sex-standardisation and via tailoring the definition of the outcome and other details of the analyses.

{kind=link}
{kind=link}
Incidence of malignancies across the five rheumatoid arthritis (RA) registry main cohorts, crude as well as age/sex-standardised to the RA trials programme, and overall crude rates as previously reported from the same registries.12–19 (A) All malignancies excluding non-melanoma skin cancer; (B) Malignant lymphomas and (C) All skin cancers. CORRONA, Consortium of Rheumatology Researchers of North America; IORRA, Institute of Rheumatology, Rheumatoid Arthritis; NOAR, Norfolk Arthritis Register; SRR, Swedish Rheumatology Quality of Care Register.
Discussion
In this study using large observational cohorts of patients with RA, we made the following observations: (1) through a concerted and harmonised approach, it was possible to define subsets of patients within the main cohorts who were more similar to RA trial populations, (2) age and sex were the main determinants of cancer incidence; RA characteristics were not strong determinants of cancer incidence, (3) taking age and sex into account, the incidences of all malignancies excluding NMSC and of malignant lymphomas were reasonably consistent across the registries, and robust across a large series of sensitivity analyses and (4) non-standardised or differently standardised rates available in the literature do not provide as tailored context as the coherent approach presented here (and certainly not patient-level data nor rates amenable to standardisation). The exception was skin cancer, which displayed too large a variation across registries and analyses to provide a readily usable benchmark for an RA trial programme.
Approval and safety contextualisation for clinical trial programmes as well as for already-licensed drugs calls for new approaches.20 Our study was set up as part of a proactive pre-approval safety programme to contextualise safety data emergent from an RA trial programme. The characteristics of this particular trial programme, the rationale for the safety contextualisation effort and challenges encountered in this, are, however, largely generic to all current antirheumatic therapies. In RA, while it is known that the overall cancer incidence is modestly elevated, it is unknown to what extent rates vary with RA characteristics or in relation to specific therapeutic exposures. Our study provides insights into these important issues, and suggests a lower level of variability across patient subsets (taking statistical precision into account) than one might have expected. This is not to say that there may not be profound differences in cancer risk across different patient segments, only that segments as defined by point measurements such as disease activity or treatment status are not, in any consistent way, major risk determinants for cancer occurrence in RA, or at least not in comparison to age and sex. The one exception is ‘all skin cancers’ which exhibited a large variation across registries. This is, however, not surprising, as rates will be reflective of the relative proportion of basal versus squamous cell skin cancer, skin type, and ultraviolet exposure and the outcome definitions (all NMSC, in situ/invasive, squamous vs basal cell) and case ascertainment methods (self-report, rheumatologist report, register-linkage) in use.21 Thus, a better understanding of skin cancer risks using RA cohorts as benchmark would require even greater attention to population and geographical characteristics, as well as a more narrowly defined and tailored outcome definition than ‘all skin cancers’.
Our study cohorts were derived from existing RA clinical cohorts/registries. The observed baseline differences in RA characteristics are a consequence of the nature of the underlying data collections more than of any geographical differences in RA phenotype and management. For instance, SRR and NOAR are early RA/arthritis cohorts, whereas CORRONA and IORRA are registries of prevalent patients.
Clearly, there are limitations when observational data are used to provide context for randomised clinical trial data. These include differences in patient populations, intensity and the length of follow-up, data availability, and definitions of variables and outcomes. In our study, limitations include the following: (1) the major established registries contributing the bulk of the data may not be fully representative of the global incidence of cancer in RA; (2) the data sources, patient population coverage, outcome variable availability and quality of data collection varied across registries; (3) data collection methods for events varied across registries, for example, from systems unrelated to the registry (linkage to administrative hospitalisation data for SRR and NOAR), via active collection from patients then validated with medical records (eg, IORRA), to data collected actively by the physician and adjudication of selected events (eg, CORRONA); (4) there is a risk of under-reporting fatal or serious outcomes if patients leave the registry or are not adequately followed up. This may be relevant for malignancy outcomes for registries without automatic linkage to cancer or hospital discharge registries (here, CORRONA, and IORRA), whereas malignancy and vital status data from SRR and NOAR would be expected to be more complete; (5) a clinical trial programme may be enriched for both ‘healthier’ and ‘sicker’ participants based on the inclusion and exclusion criteria, which may have implications for the short-term to medium-term risk of cancer. On the other hand, trial participants undergo more thorough testing and follow-up than would be experienced by patients in registries. Therefore, earlier and subclinical malignancy may be more likely to be identified in clinical trial populations, leading to an apparent higher risk for certain outcomes, at least in the short term and (6) typical Phase II/III RA trial programmes tend to have double-blind periods of less than 1 year, and even long-term extension studies on active drug provide follow-up times which, from a cancer biology point of view, are short. We addressed this issue by performing sensitivity analyses restricting the follow-up time in the registry cohorts to 18 months (instead of an average of a few years), though without noting any major effects on cancer rates.
While the extent of these biases cannot be fully known, we tried to address them with a variety of sensitivity analyses. The consistency of results across the analyses lends weight to the overall findings. Indeed, our collaborative approach has some particular strengths: a coherent approach across existing and new registries as well as direct access to patient-level data enabled us to standardise rates to a common standard as opposed to reliance on overall rates published in the literature and the possibility to tailor appropriate study populations and support sensitivity analyses and to harmonise the outcome definitions. With a main analysis that used a relatively unselected cohort over long follow-up from each registry, supplemented with a series of sensitivity analyses, we could both maximise precision across registries and provide an assessment of potential ‘bias’ in the main analysis. The selection of registry sub-cohorts allowed us to explore the effect of trying to resemble clinical study populations. Finally, temporal matching to the clinical trial programme minimised differences due to changes in risk panorama or treatment patterns over time.
In conclusion, a consistent methodology and age/sex standardisation demonstrated that, in RA populations from clinical practice from different countries and with variations in RA management and comorbidity, rates of overall malignancy excluding NMSC, solid malignancy and malignant lymphoma were reasonably consistent across registries and for within-registry sensitivity analyses. By contrast, data on all skin cancers demonstrated a larger variability and could not be as easily harmonised. Enriching information from clinical trials programmes with contextual observational data from clinical registries is a powerful means to improve the understanding of the safety profile of new drug compounds in the preapproval phase. These contextual data may also serve as a critical starting point for proactive post-approval risk management.
Acknowledgments
NOAR is funded by Arthritis Research UK (Grant reference 20380). SRR has or has had research agreements with Abbvie, Pfizer, BMS, UCB, Merck, AstraZeneca, Sobi and Roche. IORRA is supported by various grants from a large number of pharmaceutical companies, including AstraZeneca. CORRONA has received funding in the past 2 years from AbbVie, Amgen, AstraZeneca, Janssen, Genentech, Lilly, Novartis, Pfizer, Regeneron, Vertex and UCB through contracted subscriptions to the database, and CORRONA International LLC has received funding from AstraZeneca.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online tables
Footnotes
Handling editor Tore K Kvien
Contributors All listed authors fulfil the International Committee of Medical Journal Editors recommendations for authorships, and have seen and approved the final version of the manuscript.
Funding AstraZeneca funded this study (here: largely remuneration for analyst time) and collaborated with the researchers in the design, analysis and interpretation. This manuscript was produced without the use of a commercial medical writing and editorial service.
Competing interests FN, NB, TNT, EW-vS and MeHo are employees of AstraZeneca (TNT of MedImmune). FN, NB, LH, TNT, EW-vS, MeHo, SF hold AstraZeneca stocks and/or options. LH and SF were employees of AstraZeneca during the time in which the research was conducted, and CG was affiliated with the Arthritis Research UK Centre for Epidemiology. JA has had grant/research support from AstraZeneca and has been a consultant for AstraZeneca, and has received grant support from Abbvie, Pfizer, Merck, Roche, BMS and UCB. TF has received honoraria for advisory board participation from Pfizer. MH has nothing to disclose. JDG is a shareholder of CORRONA and has consulted for AstraZeneca, CORRONA, Novartis, Genentech, Regeneron and Pfizer. DAP is an employee of CORRONA and has been a paid instructor for Novartis. GR is an employee of CORRONA. KM has grant/research support from American College of Rheumatology Research Foundation and is co-director of the National Data Bank for Rheumatic Diseases (NDB), which has received funds from AstraZeneca. HY leads the IORRA cohort and has been a consultant for AstraZeneca and holds research grants from Abbvie, Astellas Chugai, Daiichi-Sankyo, Eisai, Mitsubichi-Tanabe, Santen, Taisho-Toyama, Takeda and UCB. ET has received lecture fees or consulting fees from Abbvie, Eisai Pharmaceutical, Chugai Pharmaceutical, Bristol Myers Squibb, Astellas Pharmaceutical, Pfizer, Takeda Pharmaceutical and Santen Pharmaceutical. EI has nothing to disclose. DS has had grant/research support from AstraZeneca and has been a consultant for AstraZeneca. SF, CG and LH: During the conduct of this study, SF and LH were employees of AstraZeneca, and CG was affiliated with the Arthritis Research UK Centre for Epidemiology. SV has nothing to disclose.
Ethics approval All registries had appropriate ethical approvals and patient consents.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All results are contained within the manuscript. Access to raw data will follow the principles at each of the participating institutions. The general rule is that raw data (here: the entire registers) resides with the principal investigators/governing boards of these registers, and that access to linked data will follow the legislative framework in each country.