Article Text

Abstract
Background Anti-citrullinated protein antibodies (ACPAs) are the hallmark of rheumatoid arthritis (RA). Protein citrullination is believed to drive autoantigen selection in RA. Nonetheless, several autoantigens in RA are targeted as native (unmodified) proteins. Here, the study of hnRNP A2/B1 (RA33) provides a framework to understand the humoral response to native and citrullinated autoantigens in RA.
Methods RA synovial fluid (SF) cells were analysed by immunoblotting and mass spectrometry. RA33 was cloned from RASF cells and splice variants expressed as recombinant proteins. Antibodies against native and citrullinated RA33 were characterised by ELISA, immunoblotting and immunoprecipitation.
Results RA33 is citrullinated in the rheumatoid joint and targeted either as a citrullinated or native protein in distinct patient subsets with RA. A novel splice variant (hnRNP B1b) previously associated with disease initiation in experimental arthritis was identified in the RA joint and acts as the major target of the anti-RA33 response. Antibodies exclusively targeting citrullinated RA33 were positively associated with disease duration and erosive disease. In contrast, anti-(native) RA33 antibodies were detected almost exclusively in early RA and identified patients with low radiographic erosion scores. Finally, a unique subset of double-reactive patients demonstrated intermediate severity, but rapid disease progression, suggesting a transitional disease phase in the evolution of an anti-native protein antibody to ACPA response in RA.
Conclusions These data suggest that native and citrullinated proteins targeted by autoantibodies in RA may be part of a single antibody system and challenge the paradigm of citrullination as the unifying principle underlying loss of tolerance in RA.
- Rheumatoid Arthritis
- Ant-CCP
- Autoantibodies
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease of unknown aetiology characterised by a high-titer antibody response against self-antigens. The discovery that patients with RA have antibodies against citrullinated proteins (ACPAs) marked an important advance in elucidating a unifying principle that may explain autoantigen selection in this disease.1 In this model, loss of tolerance to peptidylcitrulline is thought to drive immune events that initiate and propagate the disease.2 While the discovery of ACPAs has critically aided early diagnosis and propelled the study of immunopathology in RA,3 ,4 it has inherently dwarfed interest in understanding autoantibody systems, which specifically recognise native (ie, unmodified) autoantigens in RA. This family of autoantigens includes Fc-gamma (targeted by rheumatoid factor, RF), hnRNP A2/B1 (RA33), calpastatin and peptidylarginine deiminase type 4 among others.5–9
Anti-native protein autoantibodies are frequently detected in both ACPA-positive and ACPA-negative RA9–12 and may distinguish clinical subsets within these patient populations.11–14 How the autoantibody systems targeting native and citrullinated proteins in RA are mechanistically related is unknown. Intriguingly, experimental models of arthritis have not been able to validate citrullination as a major determinant of antigenicity15–19 and fail to mount measurable ACPA responses.16–20 Nonetheless, these animals distinctly target RA autoantigens in their native (unmodified) form, including antigens that are recognised by ACPAs in RA (vimentin, filaggrin, apolipoprotein A-I, and histones).15 ,18–20 While supporting the significance of a common set of autoantigens for both RA and experimental arthritis, these findings question whether citrullination represents the unifying principle that explains the generation of an autoimmune response to arthritogenic autoantigens. Here, we study RA33 to understand the dual autoantibody responses to native and citrullinated proteins in RA.
Materials and methods
Study participants
Serum was obtained from 196 patients with established RA recruited as part of a longitudinal cohort (ESCAPE RA).21 Disease activity and severity were assessed at baseline and at two additional time points, with the final visit occurring an average of 39±4 months after enrolment. Serum from 56 healthy donors served as controls. All individuals provided informed consent as approved by the Johns Hopkins Institutional Review Board (IRB). Synovial fluid (SF) was obtained after clinically indicated arthrocentesis under a Partners Healthcare IRB–approved protocol.
Cloning, recombinant expression and antigenic characterisation of RA33
RA33 splice variants were cloned from RA SF and expressed as recombinant proteins. Citrullination sites were identified by mass spectrometry (MS) as previously described.22 Native and citrullinated RA33 were used to detect autoantibodies in RA and healthy donor serum by immunoblotting, immunoprecipitation (IP) and ELISA. Detailed descriptions are available in the online supplementary methods.
Results
RA33 is citrullinated in the rheumatoid joint
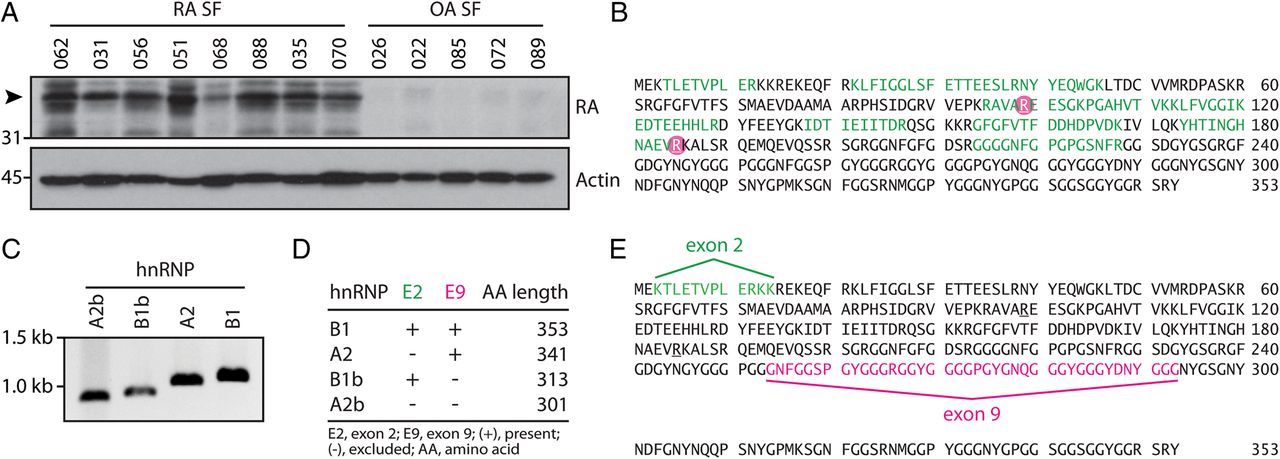
SF cell lysates from patients with RA and osteoarthritis (OA) were analysed by immunoblotting and probed with ACPA-positive RA serum to identify unique autoantibody specificities in the rheumatoid joint. A prominent ∼38 kDa band detected in RA, but not in OA SF cells (figure 1A), was further analysed by MS. hnRNP A2/B1 (RA33) and hnRNP A3 were identified as candidate citrullinated RA autoantigens that matched the migration pattern observed by immunoblotting (see online supplementary table S1). RA33 was described as an autoantigen almost 10 years before the discovery of ACPAs1 ,23 and identified as a common target of autoantibodies in both RA (15–35%) and experimental arthritis.16 ,17 ,23–25 Unlike the erosive disease associated with ACPAs and RF,14 anti-RA33 antibodies have been uniquely associated with a milder, non-erosive disease phenotype.11 Citrullination of RA33 at arginine residue 99 (R99) was confirmed in independent RA SF cell samples (Ascore 1000) (figure 1B and see online supplementary table S1). In addition, published data sets of the extracellular RA SF proteome identified a second citrullination site of RA33 at arginine residue 185.26 Given the previous identification as a native antigen in RA and its primary role for disease initiation in experimental arthritis,16 ,17 ,27 we opted to further characterise citrullinated RA33 as a candidate autoantigen in RA.
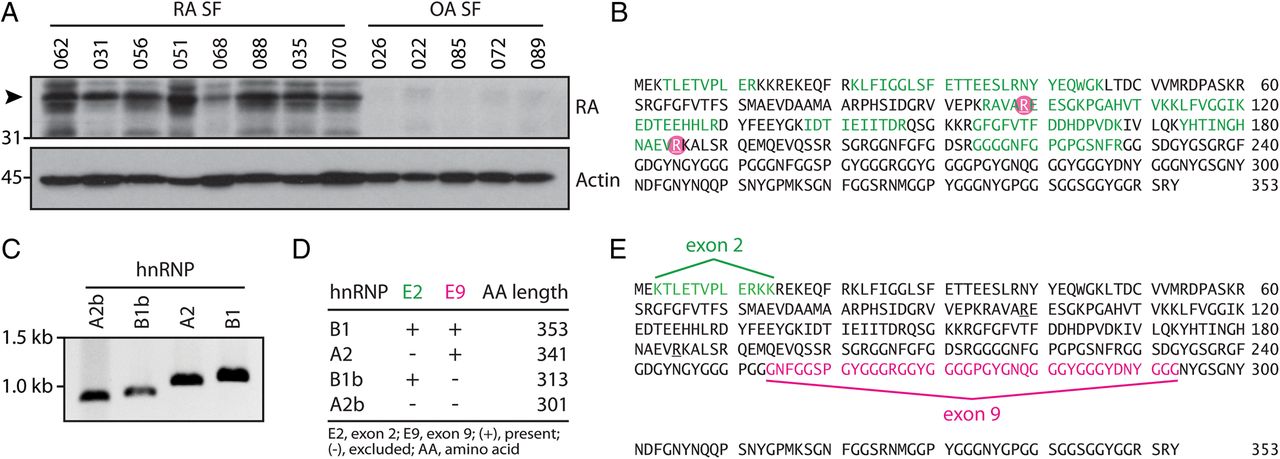
Alternative splicing and posttranslational modification of RA33 (hnRNP A2/B1) in the rheumatoid joint. (A) Synovial fluid (SF) cells from patients with rheumatoid arthritis (RA SF) and osteoarthritis (OA SF) were analysed by immunoblotting using anti-citrullinated protein antibody (ACPA)-positive RA patient serum. Data from one representative serum detecting a ∼38 kDa band in RA SF cell samples is shown (top panel). Detection of actin is shown as a loading control (lower panel). (B) Citrullination sites of hnRNP A2/B1 (RA33) as identified by mass spectrometry (MS) analysis in RA SF cells and supernatant26 are circled in pink. Peptide sequence coverage is shown in green. (C) Novel splice variants of RA33 are expressed in the RA joint. PCR of major (hnRNP A2/B1) and minor (hnRNP A2b/B1b) splice variants of RA33 cloned from RA SF cDNA. (D) Alternative exon usage of RA33 variants expressed by RA SF cells as determined by Sanger sequencing. Expected protein lengths of all four identified variants are shown. (E) Splicing map of RA33 in relation to the full amino acid sequence of hnRNP B1. Sequences excluded by alternative splicing in hnRNP A2 and hnRNP A2b (exon 2) are highlighted in green, and exon 9-encoded sequences excluded in b-variants of RA33 are highlighted in pink.
To study citrullinated RA33, we cloned cDNA encoding hnRNP A2/B1 from RA SF cells. Four transcript variants encoded by the same gene were identified, designated hnRNP A2, B1, A2b and B1b (figure 1C). hnRNP B1 is the full transcript variant, whereas in hnRNP A2, exon 2 is excluded by alternative splicing. The corresponding proteins are almost identical, with the exception of 12 amino acids (positions 3–14) that are absent in hnRNP A2 (figure 1D, E).16 hnRNP A2b and B1b are short variants of hnRNP A2/B1 that result from alternative splicing of exon 8 to exon 10, skipping exon 9 (figure 1D, E). Intriguingly, these short variants have been identified in the synovium of animals with experimental arthritis16 but are generally undetectable in most human tissues.28 We next expressed all variants as recombinant proteins. Only minor differences in citrullination by PAD4 were observed between the four splice variants by anti-modified citrulline immunoblotting (figure 2A). Using MS, citrullination was confirmed in 16/25 arginine residues of citrullinated hnRNP A2 (citA2) and citrullinated hnRNP B1 (citB1) (figure 2B, C), including residues 99 (R99) and 185 (R185) identified in the rheumatoid joint. R12 in the exon 2-encoded region unique to B1 and R226 in A2 were the only citrullination sites that differed among these variants (figure 2B, C, respectively).
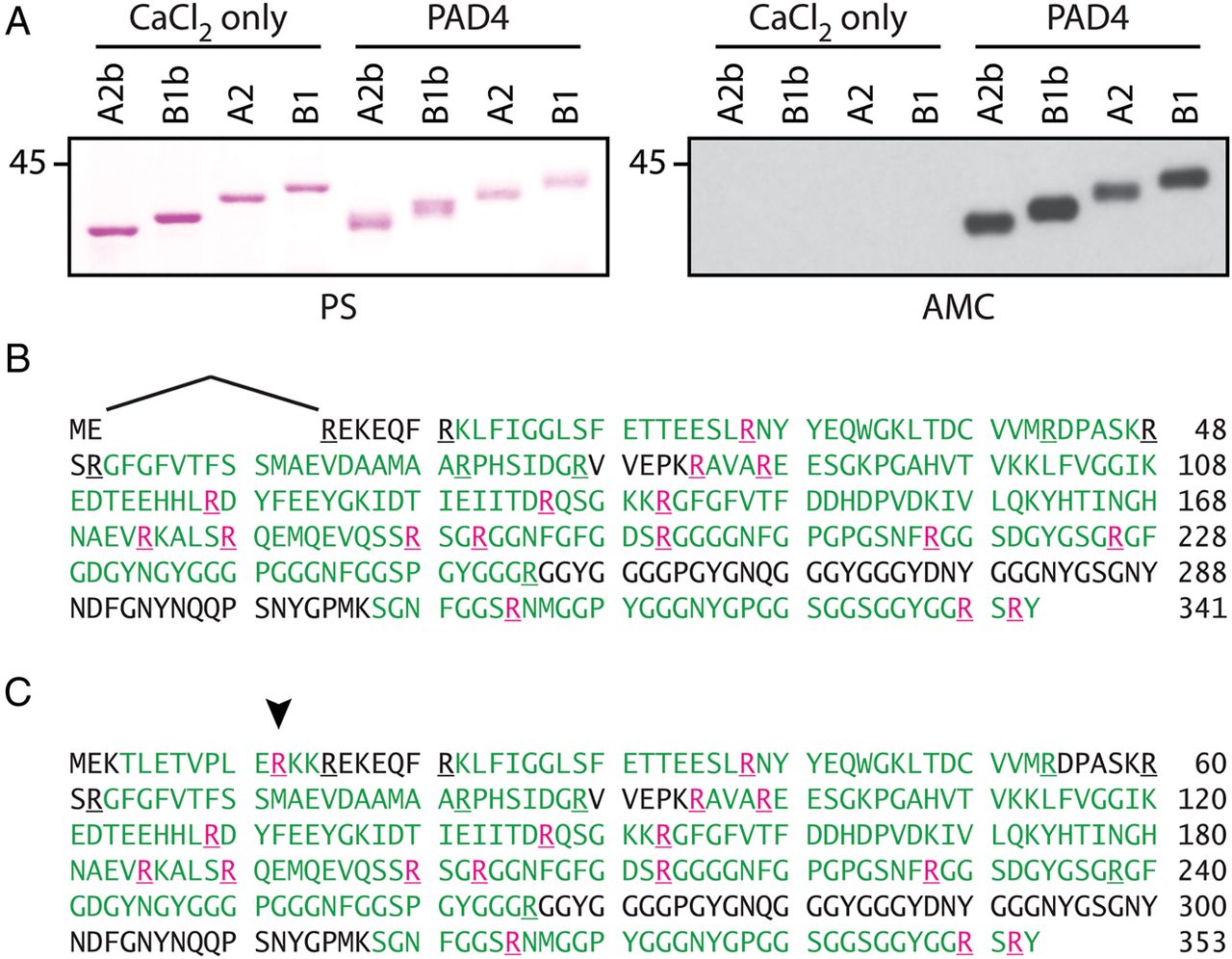
Citrullination of purified recombinant RA33 by PAD4. (A) Anti-modified citrulline (AMC) immunoblotting of native and citrullinated hnRNP A2b, B1b, A2 and B1. Purified splicing variants were incubated in the absence or presence of PAD4 (right panel). Ponceau staining (PS) is shown to visualise loading (left panel). (B and C) Mass spectrometry (MS) sequence coverage map of in vitro citrullinated hnRNP A2 (B) and B1 (C). Peptide sequences identified by MS are shown in green; citrullination sites in pink (localisation probability >98% for each citrulline residue shown). The arrowhead indicates R12 citrullination in the exon 2-encoded region unique to B1.
RA33 is targeted as a citrullinated autoantigen in RA
Using in vitro citrullinated and native hnRNP A2, B1, A2b and B1b, we examined how splicing and/or citrullination affect antibody binding in RA. We initially screened 18 anti-cyclic citrullinated peptide (CCP) antibody positive patients for antibodies against native and citrullinated hnRNP A2b and B1b by ELISA (figure 3A). Antibody reactivity was significantly increased when using citrullinated versus native RA33 (mean 0.076 vs 0.015; p=0.004 and 0.197 vs 0.014; p<0.0001 for citA2b vs A2b and citB1b vs B1b, respectively). Next, we compared the reactivity of the same patient sera against citA2, citB1, citA2b and citB1b (figure 3B and see online supplementary figure S1). Anti-citRA33 antibodies showed significantly increased reactivity against citB1b vs citA2b (mean 0.197 vs 0.076; p<0.0001). Analogously, reactivity against citB1 was significantly higher than against citA2 (mean 0.125 vs 0.043; p<0.0001), indicating that B1 and B1b are preferential targets of the anti-citRA33 response in RA. Preferential antibody reactivity was moreover seen with both citB1b (mean 0.197 vs 0.125 for citB1; p=0.0004) and citA2b (0.076 vs 0.043 for citA2; p=0.0004) over their citrullinated long splice variants, suggesting that exclusion of exon 9 modifies antigenicity of the citrullinated proteins. Interestingly, carbamylation of hnRNP B1b abrogated antibody reactivity, suggesting anti-modified RA33 autoantibodies in RA are citrulline specific (figure 3C).

Antibodies to native and modified RA33 in rheumatoid arthritis (RA). (A) Antibody reactivity of anti-CCP positive RA sera (n=18) against native and citrullinated hnRNP A2b and B1b by ELISA (Wilcoxon-matched pairs test). (B) Serum reactivity against citrullinated hnRNP A2 (citA2), citrullinated hnRNP B1 (citB1) and their citrullinated variants hnRNP A2b (citA2b) and hnRNP B1b (citB1b) in anti-CCP positive RA (n=18) by ELISA. Wilcoxon-matched pairs test (***p<0.001 and ****p<0.0001). (C) Comparison of antibody reactivity against native (B1b), carbamylated (carbB1b) and citrullinated (citB1b) RA33 in anti-CCP positive RA. Friedman’s test (*p<0.05 and ****p<0.0001). (D) Antibodies against native RA33 (B1b) and citrullinated RA33 (citB1b) in patients with established RA (n=196) and healthy subjects (n=56) by ELISA. Black lines represent mean reactivity; dotted lines mark the 95th percentile of controls. Antibody reactivity is expressed in arbitrary units (AU). ns, Not significant.
Antibodies to native and citrullinated RA33 identify distinct patient subsets that differ in disease duration and phenotype
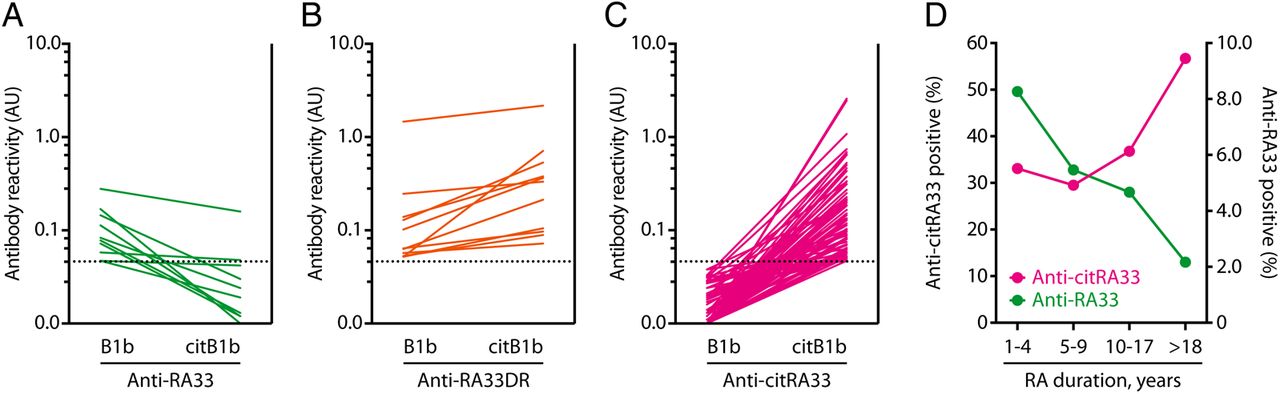
To establish the prevalence and significance of antibodies against native and citrullinated RA33, we assayed serum of 196 patients with RA and 56 healthy controls for anti-B1b and anti-citB1b by ELISA (figure 3D). Antibodies against citB1b (anti-citRA33) were identified in 86 (43.9%) patients with RA, whereas anti-native B1b antibodies (anti-RA33) were detected in 21 (10.7%) patients. Levels of anti-citRA33 were significantly higher in RA versus controls (mean 0.133 vs 0.013; p<0.0001). Among patients with RA, anti-citRA33 vs anti-RA33 levels were significantly higher (mean 0.133 vs 0.029; p<0.0001) (figure 3D). RA33 was targeted exclusively as a citrullinated antigen in the majority of antibody-positive patients (75/96, 78%; 75/196, 38% of all RA) (figure 4C), while antibodies specific to native RA33 were identified in 10 patients (10/96, 10%; 10/196, 5% of all RA) (figure 4A). A unique subset of patients (11/96, 11%; 11/196, 6% of all RA) was found to target both native and citrullinated RA33 (anti-RA33 double reactive RA (anti-RA33DR)) (figure 4B).

Antibodies to native and citrullinated RA33 follow opposing patterns of positivity in early and late rheumatoid arthritis (RA). (A–C) Reactivity of individual patient with RA sera against native and citrullinated hnRNP B1 by ELISA. Patients with RA target RA33 in three distinct ways: (A) exclusively as a native antigen (green); (B) both as a native and citrullinated antigen (orange) and (C) exclusively as a citrullinated antigen (pink). Sera with preferential binding to native hnRNP B1b were classified as native specific. Dotted lines mark cut-offs for positivity based on the 95th percentile of controls. (D) Frequency of anti-citRA33 antibody positivity (pink circles) and anti-RA33 antibody positivity (green circles) in patients with RA grouped by disease duration (1–4, 5–9, 10–17 and >18 years).
In accordance with a previous study,25 we found that anti-RA33 antibodies were more frequently observed in patients with shorter disease duration (16.4% of patients <4 years after disease onset), and progressively lower in patients later in their disease course (6.3% of patients at >18 years after disease onset). This trend was inversely observed for anti-citRA33 antibodies, which were more prevalent in patients with longer standing RA (56.3% at >18 years) (figure 4D). The identification of distinct patient subsets targeting RA33, citRA33 or both warranted us to re-address the clinical significance of these antibodies. Anti-RA33 antibodies identified patients with the lowest systemic inflammatory burden and with erosion scores similar to patients negative for these antibodies (see online supplementary table S2). This is consistent with the attenuated disease previously reported in these patients.11 In contrast, patients with anti-citRA33 antibodies (anti-RA33 negative) demonstrated markedly increased levels of radiographic damage compared with seronegative patients (median total Sharp/van der Heijde (SvdH) score=17 vs 5 units, respectively; p=0.006, and median total erosion score=7 vs 2 units, respectively; p=0.001), despite equivalent therapeutic intervention (see online supplementary table S2). Similarly, patients with anti-citRA33 vs anti-RA33 antibodies tended to exhibit higher markers of erosive disease (median total erosion score 7 vs 1.5; p=0.07), although this analysis was limited by sample size.
While anti-RA33 antibodies and anti-citRA33 antibodies identified patient subsets at opposite ends of the clinical spectrum, patients with antibodies against both RA33 and citRA33 were characterised by intermediate disease severity, but rapid radiographic progression. Patients with anti-RA33DR antibodies had median disease duration of 6 years as compared with 13 years in anti-citRA33 single positive RA (anti-citRA33 SP) (figure 5A), suggesting that this pattern of reactivity is observed earlier in the disease course. Patients with anti-RA33DR antibodies showed higher erosive burden than anti-RA33 single positive RA (anti-RA33 SP) (median SvdH score=12 vs 6 units, respectively), but less than anti-citRA33 SP (median SvdH score=12 vs 17 units, respectively) (figure 5C). Similarly, the median ACPA number in anti-RA33DR RA was markedly higher compared with anti-RA33 SP (5 vs 0; p=ns), but lower than in patients with anti-citRA33 SP (5 vs 7; p=ns) (figure 5B). However, sample size was prohibitive to reach statistical significance. Strikingly, patients with anti-RA33DR antibodies had the highest rate of radiographic progression (median ΔSvdH score/year 2.3 vs 0 units/year in anti-RA33 SP; p=0.025, and 2.3 vs 0.6 units/year in anti-citRA33 SP; p=0.036) (figure 5D).

Anti-RA33DR autoantibodies identify patients with a transitional phenotype and accelerated disease progression. (A) Median rheumatoid arthritis (RA) disease duration (years) and median patient age (years) based on antibody status (anti-RA33 SP, anti-RA33DR and anti-citRA33 SP). (B) Median anti-citrullinated protein antibody (ACPA) number and anti-CCP antibody positivity (in %) in RA based on antibody status. (C) Median Sharp/van der Heijde (SvdH) score and joint space narrowing (JSN) score in RA based on antibody status. (D) Median change in SvdH score per year and median circulating IL-6 levels in the three RA33 reactive groups.
RA33 is distinctly targeted as a native and citrullinated autoantigen in RA
Several ACPA specificities were initially identified as reactivities against the native protein, including α-enolase,29 vimentin (Sa),30 type II collagen31 and actin.32 Interest in further characterisation of these native targets, however, has been surpassed by their study as citrullinated antigens. Whether binding to the native antigen is the result of ACPA cross-reactivity or indeed mediated by a different subset of autoantibodies is unclear. The finding that antibodies against RA33 have distinct patterns of antigen binding (dependent vs independent of antigen citrullination) offers an opportunity to reassess the determinants of autoantibody recognition in RA.
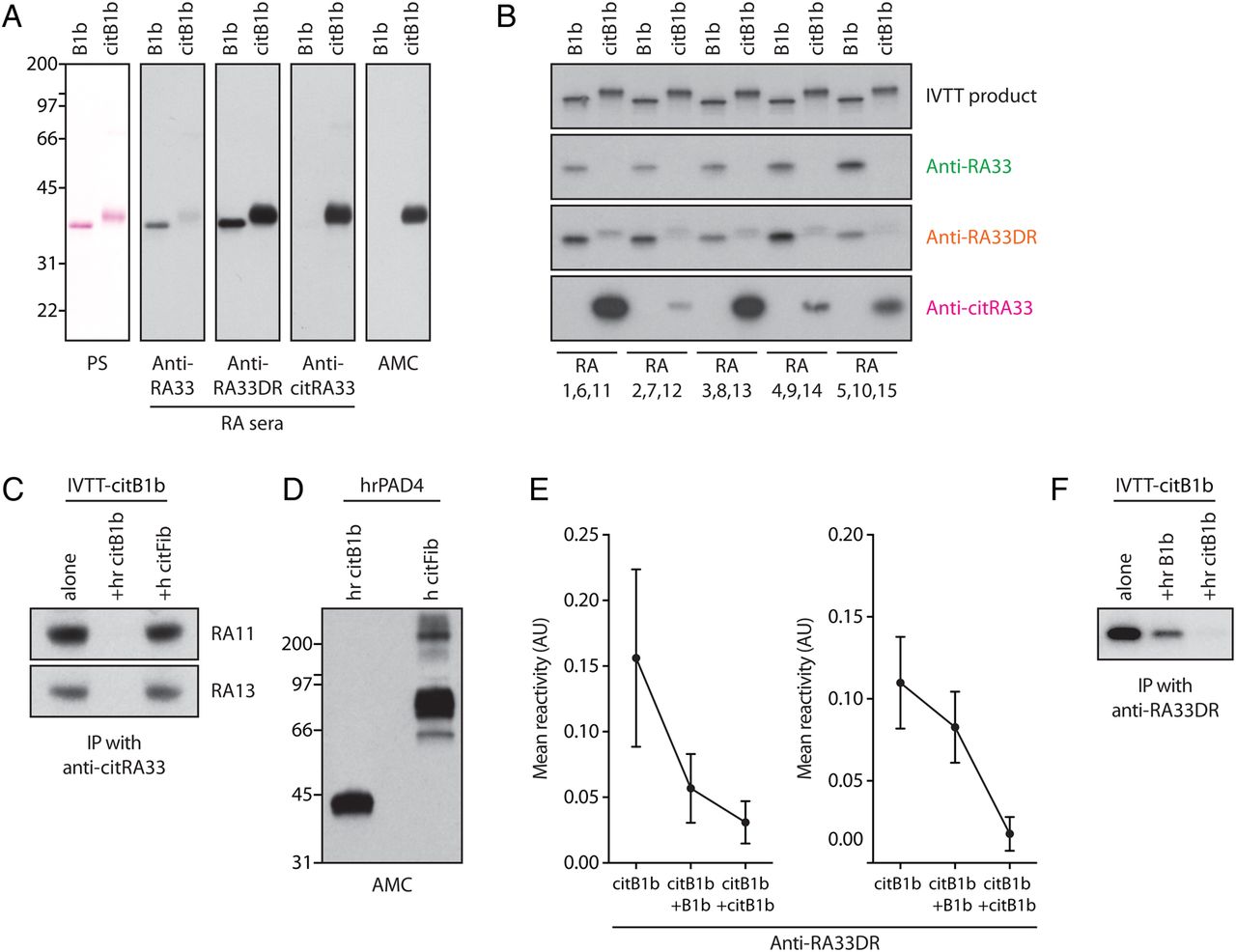
We further characterised the anti-RA33 response using two independent methods (immunoblotting and IP using radiolabelled in vitro transcribed–translated (IVTT) protein). Patterns determined by ELISA were equally observed by immunoblotting (figure 6A) and by IP of native and citrullinated IVTT-RA33 (figure 6B), confirming the distinct antibody types identified. By IP, the reactivity of anti-RA33 SP sera with native IVTT-RA33 was blocked by coincubation with recombinant RA33, but fully retained by incubation with recombinant citRA33 (data not shown), suggesting that anti-RA33 SP represent a population of true anti-native protein antibodies in RA. Moreover, the reactivity of anti-citRA33 SP sera with IVTT-citRA33 was abrogated by recombinant citRA33, but unaffected by citrullinated human fibrinogen, strongly supporting that the reactivity against citRA33 is specific and not the result of ACPA cross-reactivity (figure 6C, D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RA33 is distinctly targeted as a native and citrullinated autoantigen in rheumatoid arthritis (RA). (A) Western blot for native (B1b) and citrullinated hnRNP B1 (citB1b) using representative RA sera specific for RA33, citrullinated RA33 (citRA33) or both (RA33DR) as identified by ELISA (panels 2–4). Ponceau staining (PS) is shown to visualise loading (panel 1), and citrullination was detected by anti-modified citrulline (AMC) immunoblotting (panel 5). (B) Immunoprecipitation of radiolabelled in vitro transcribed–translated (IVTT)-B1b and IVTT-citB1b using RA patient sera representative of the three groups of anti-RA33 positivity (panels 2–4). Native and citrullinated IVTT products directly resolved by SDS–PAGE are shown to demonstrate loading. The shift in molecular weight seen for citB1b indicates citrullination (top panel). (C) Competition assay/immunoprecipitation (IP) of radiolabelled IVTT-citB1b by two sera positive for anti-citRA33 antibodies in the absence (alone) or presence of human recombinant citrullinated B1b (+hr citB1b) or human citrullinated fibrinogen (+hr citFib). (D) AMC immunoblot of in vitro citrullinated hr B1b and citrullinated fibrinogen used in competition assays. (E) Competition assays interrogating the reactivity of anti-RA33DR sera against citrullinated RA33 by ELISA. Graphs show reactivity of anti-RA33DR sera against citB1b after preincubation with buffer alone, native B1b or citB1b (mean reactivity±SEM). Patterns of inhibition demonstrate varying degrees of cross-reactivity (shown in the left and right graphs). (F) IP of radiolabelled IVTT-citB1b by one representative anti-RA33DR serum in the absence (alone) or presence of recombinant native B1b (+hr B1b) and citrullinated B1b (+hr citB1b).
Anti-RA33DR antibodies are of particular interest as they may represent a link between anti-RA33 SP and anti-citRA33 SP antibodies. Using an ELISA-based approach, the reactivity of some anti-RA33DR sera against citRA33 was similarly inhibited by preincubation with both native and citrullinated RA33 (figure 6E, left graph), suggesting these sera contain predominantly cross-reactive antibodies. In other patients, antibody binding was abrogated by citrullinated RA33, but only partially inhibited by the native protein (figure 6E, right graph), indicating that these patients harbour additional monospecific antibodies. These patterns were similarly observed when testing individual anti-RA33DR sera by IP (figure 6F), where a fraction of citRA33 binding was inhibited by the unmodified antigen. These assays suggest that anti-RA33DR sera contain a heterogeneous population of cross-reactive and monospecific antibodies against native and citrullinated RA33.
Together, these data demonstrate that RA33 is distinctly targeted as native and as citrullinated antigen in RA. Moreover, they support a clinical stage in which cross-reactive and antigen-specific antibodies co-exist, suggesting a stage of transition between the anti-native and anti-citrullinated protein responses in patients with RA.
Discussion
The humoral response in RA is remarkably diverse. It is characterised by the presence of different antibody systems that target native proteins as well as proteins containing posttranslational modifications (PTMs).1 ,33 ,34 While current models of RA have embraced PTMs as core principles of pathogenesis,2 ,4 ,34 the occurrence of anti-native protein antibodies is not adequately explained by the PTM-centric paradigm of autoantigen selection in RA.
Among RA autoantigens, RA33 is of particular interest due to its relevance as an unmodified protein target in both human and animal models of arthritis.16 ,17 ,23 In experimental arthritis, RA33 is strikingly overexpressed in the joint of human tumour necrosis factor (TNF)-α transgenic (hTNFtg) mice and pristane-induced arthritis (PIA), which coincides with the appearance of anti-RA33 antibodies and precedes the onset of symptomatic disease.16 ,17 Overexpression includes cryptic variants of RA33 (hnRNP A2b/B1b),16 whose human orthologues we identified in the RA joint. De novo expression of these variants has been linked to loss of tolerance in hTNFtg mice16 and may be similarly relevant in RA. Importantly, ACPAs are not detected in hTNFtg mice or PIA,16 ,17 ,19 illustrating that citrullination is not essential for the development of anti-RA33 antibodies. In contrast, RA33 expression and antibodies were closely linked to TNF-α signalling in these models,16 ,27 suggesting that loss of tolerance to this inducible synovial antigen may be driven by TNF (rather than by antigen citrullination).
In patients with RA, anti-RA33 antibodies have been shown to target hnRNP A2/B1 as an unmodified protein,6 ,25 which may suggest that RA33 is a distinct antibody system in the aetiopathogenesis of RA, presumably driven by mechanisms independent of citrullination. Our findings challenge this interpretation by identifying RA33 as citrullinated synovial protein that is targeted by ACPAs. The analysis of fine specificities recognised by RA33 antibodies in RA demonstrated that RA33 is indeed targeted in three ways: (1) exclusively as a citrullinated antigen, (2) both as a native and citrullinated antigen and (3) exclusively as a native antigen.
Two alternative models are conceivable to explain the humoral response against native and citrullinated RA33 in RA. Based on the current model of RA, in which autoantigen selection is driven by the generation of citrullinated neoepitopes (ie, a citrulline-centric model), citrullination of RA33 would initiate loss of tolerance and antibody production. While anti-citrullinated RA33 antibodies are perfectly consistent with this model, an antibody response against only the native protein cannot easily be derived. The fact that RA33 is conserved as a major native antigen in RA and ACPA-negative experimental arthritis6 ,16 ,17 and the bimodal distribution of antibodies against native and citrullinated RA33 observed in this study suggest an alternative model in which the antigen is initially targeted independent of citrullination. In this model (antigen-centric model), RA33 would be preselected as a native autoantigen and only later targeted as a citrullinated protein. Several lines of evidence from this and other studies support an antigen-centric model of antigen selection over the classic citrulline-centric paradigm in RA.
First, the transition of mild, non-erosive arthritis associated with anti-RA33 antibodies to advanced erosive disease is characterised by the appearance of antibodies that target citRA33, suggesting that protein citrullination may augment, rather than initiate, the autoimmune response that drives RA. Second, assuming antibody evolution from an exclusively native pattern of antigen recognition to a humoral response that exclusively targets the citrullinated antigen, we would expect to find evidence that both antigens may be targeted simultaneously at one point in time. This would coincide with a transition in disease phenotype. The phenotypically distinct population of patients with anti-RA33/citRA33 DR antibodies described herein may identify this transitional state in the subset of established RA that still evolves to ACPA-positive disease after diagnosis. Third, a recent study reported that reactivity against native peptides derived from RA autoantigens was detectable in a subset of presymptomatic RA (pre-RA),35 where the median time of first positivity preceded that of the matched citrullinated peptides. As is the case with our cross-sectional findings, these data do not reflect antibody evolution in individual patients but show that antibodies against native peptides may occur prior to their anti-citrullinated peptide counterparts. Finally, an interesting study by de Pablo and colleagues showed that patients with periodontal disease, a possible trigger of RA, have a predominant antibody response against uncitrullinated peptides of RA autoantigens,36 suggesting that any evolution from periodontitis to pre-RA may start with a preselected antibody response against native antigens. This has been similarly suggested for patients with bronchiectasis,37 a population that may include individuals at risk to develop RA. The idea that ACPAs are directed against preselected antigens may explain why only a subset of citrullinated proteins is actually targeted by ACPAs, despite a plethora of citrullinated proteins in hypercitrullinated cells and target tissues.38
Applying the antigen-centric model, the transition of an anti-native protein to an anti-citrullinated protein antibody response may depend on the presence of permissive factors, that is, a genetic predisposition allowing for presentation (HLA alleles) and the continued production of modified antigen (chronic bacterial infection, smoking or immune-mediated membranolysis resulting in secondary release of modified autoantigens into the extracellular compartment of the RA joint).4 ,36 ,38 ,39 ,40 In the individual at risk, concurrence of these factors, timing and stochastic events may define whether antibody transition occurs during the pre-RA phase, once the disease is established, or not at all (ACPA-negative RA). In experimental arthritis, it is possible that the transition from anti-native to anti-citrullinated protein antibody response may not occur (or occur only late), explaining why the antibody repertoire in these ACPA-negative models of experimental arthritis is limited to RA autoantigens in their unmodified form.18–20
Anti-native protein antibodies may represent markers of the earliest disease phase in the pathogenesis of RA, preceding the development of the ACPA response. As such, they present a largely unexplored opportunity to study specific populations at risk (eg, patients with periodontal disease) and to elucidate mechanisms of antigen selection in existing models of experimental arthritis, which target citrullinated RA autoantigens exclusively as native proteins.
Acknowledgments
The authors thank Lauren DeVine and Kevon Sampson for technical assistance and support, Joan M Bathon (Columbia University) for providing valuable RA serum samples and Jeremy Sokolove (Stanford University) for providing analyses of ACPA number and repertoire.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors Each author has contributed to one or more of the following aspects of the manuscript—conception and design, acquisition, analysis and interpretation of data, drafting and revising the article.
Funding This work was supported by The Jerome L Greene Foundation, The Donald B and Dorothy L Stabler Foundation (FA), the Rheumatology Research Foundation (JTG) and the Fundación Bechara (PAN).
Competing interests None declared.
Ethics approval Johns Hopkins Institutional Review Board, Partners Healthcare Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data relevant for this study have been included in the manuscript. Any additional data referred to as not shown will be available through direct contact with the corresponding author. Reagents derived from this project will be available upon request through material transfer agreements.