Article Text

Abstract
Objective Despite the importance of type I interferon (IFN-I) in systemic lupus erythematosus (SLE) pathogenesis, the mechanisms of IFN-I production have not been fully elucidated. Recognition of nucleic acids by DNA sensors induces IFN-I and interferon-stimulated genes (ISGs), but the involvement of cyclic guanosine monophosphate (GMP)–AMP synthase (cGAS) and stimulator of interferon genes (STING) in SLE remains unclear. We studied the role of the cGAS–STING pathway in the IFN-I-producing cascade driven by SLE serum.
Methods We collected sera from patients with SLE (n=64), patients with other autoimmune diseases (n=31) and healthy controls (n=35), and assayed them using a cell-based reporter system that enables highly sensitive detection of IFN-I and ISG-inducing activity. We used Toll-like receptor-specific reporter cells and reporter cells harbouring knockouts of cGAS, STING and IFNAR2 to evaluate signalling pathway-dependent ISG induction.
Results IFN-I bioactivity and ISG-inducing activities of serum were higher in patients with SLE than in patients with other autoimmune diseases or healthy controls. ISG-inducing activity of SLE sera was significantly reduced in STING-knockout reporter cells, and STING-dependent ISG-inducing activity correlated with disease activity. Double-stranded DNA levels were elevated in SLE. Apoptosis-derived membrane vesicles (AdMVs) from SLE sera had high ISG-inducing activity, which was diminished in cGAS-knockout or STING-knockout reporter cells.
Conclusions AdMVs in SLE serum induce IFN-I production through activation of the cGAS–STING pathway. Thus, blockade of the cGAS–STING axis represents a promising therapeutic target for SLE. Moreover, our cell-based reporter system may be useful for stratifying patients with SLE with high ISG-inducing activity.
- systemic lupus erythematosus
- inflammation
- cytokines
- autoimmune diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease in which pathogenic autoantibodies are produced against nucleic acids and their interacting proteins, resulting in inflammation and tissue damage. SLE is characterised by flares and remissions, with multiple organ damage ranging from mild rash to severe nephritis, and has a poor prognosis.1 2
Although the exact cause of SLE remains unclear, type I interferon (IFN-I) plays important roles in its pathogenesis and progression.3 Serum IFN-I and expression of multiple IFN-I-stimulated genes (ISGs) in peripheral blood mononuclear cells (PBMCs) are elevated in SLE and associated with SLE disease activity.4–6 Also, clinical lupus occasionally occurs after treatment of chronic hepatitis with recombinant IFN-α.7 Furthermore, although SLE is heterogeneous, over 80% of patients exhibit a prevalent IFN signature.8 Clinical trials revealed that IFN-I-targeting therapies are more effective in patients with high ISG expression.9 10
Activation of pattern-recognition receptors (PRRs) is important for IFN-I production. Two major PRR signalling pathways recognise DNA: Toll-like receptor 9 (TLR9) and cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING).11 12 These DNA-sensing pathways are crucial for SLE onset. A recent genome-wide association study demonstrated that several alleles of genes related to nucleic acid degradation and IFN-I signalling are associated with the risk of SLE.13–17 In humans and mice, defective DNA degradation leads to excessive DNA and systemic inflammation via overproduction of IFN-I.18–21 Also, stimulation of TLR9 by double-stranded DNA (dsDNA) and immune complexes (ICs) from patients with SLE results in IFN-I overproduction.22 23 TLR9 is important in SLE pathogenesis, but other molecular mechanisms also contribute to IFN-I production.24 25 Given that cGAS and STING are involved in DNA recognition,26–28 the cGAS–STING pathway may play a role in SLE.
Although DNaseII−/− mice die during development due to overproduction of IFN-I, both Sting−/−DNaseII−/− and cGAS−/−DNaseII−/− prevent embryonic lethality and reduce lupus-like manifestation, despite intact production of autoantibodies.29–31 In three prime repair exonuclease 1 (Trex1)-deficient mice, development of lupus-like symptoms is ameliorated by knockout of cGAS or STING.30 32 In humans, gain-of-function mutation in STING causes SLE-like disease.33 34 Furthermore, PBMCs of SLE express higher levels of cGAS, and approximately 15% of patients with SLE express detectable levels of cyclic GMP–AMP (cGAMP) in their PBMCs.35 However, the role of the cGAS–STING pathway in SLE pathogenesis remains unclear. To address this issue, we used patient samples and a cell-based reporter system that enables highly sensitive measurement of serum IFN-I bioactivity and ISG-inducing activity.
Materials and methods
Clinical samples
Subjects were Japanese patients admitted to the Department of Clinical Immunology, Osaka University, from 2012 to 2017. SLE (n=75), systemic sclerosis (SSc) (n=24) and primary Sjögren syndrome (SjS) (n=11) were diagnosed based on the 1997 American College of Rheumatology (ACR) revised criteria for the classification of SLE,36 the 1980 ACR criteria for the classification of SSc37 and the American-European Consensus Group 2002 criteria for SjS,38 respectively. Healthy control (HC) samples were obtained from 31 volunteer donors. Eleven patients were excluded from the study according to the criteria described in online supplementary figure S1. Disease activity of SLE was assessed according to Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score.39
Supplementary file 1
Reporter cell lines
THP1-Blue ISG (THP1-ISG), HEK-Blue IFN-α/β, THP1-Dual, THP1-Dual-KOSTING and THP1-Dual-KOcGAS cells were purchased from InvivoGen. Knockout lines THP1-ISG-KOSTING and THP1-ISG-KOIFNAR2 were generated from THP1-ISG using the CRISPR-Cas9 gene-editing system.40 Briefly, THP1-ISG was lentivirally transduced with lentiCas9-Blast and lentiGuide-Puro (Addgene) fused with sgRNA. Monoclonal knockout cells were confirmed by western blotting and DNA sequencing.
Cell-based reporter assay
For measurement of IFN-I, HEK-Blue IFN-α/β was incubated with 20% serum. For measurement of ISG-inducing activity, phorbol 12-myristate 13-acetate (PMA)-treated THP1-ISG was cultured with 20% serum. Secreted embryonic alkaline phosphatase (SEAP) in supernatant was detected with QUANTI-Blue (InvivoGen) and quantified by optical density at 620 nm. PBMCs from healthy donors were isolated with Ficoll-Paque Plus (GE Healthcare). PBMCs were incubated with 20% serum, and IFN-I levels in culture supernatant were measured with HEK-Blue IFN-α/β. For isolation of apoptosis-derived membrane vesicles (AdMVs), serum was centrifuged at 1700 g for 15 min to remove cell debris, then centrifuged twice at 16 000 g for 20 min at 4°C, and pellets were reconstituted in culture medium or phosphate buffered saline (PBS).
Extraction and measurement of dsDNA
Extracellular dsDNA in serum or AdMVs was extracted using DNA Extractor SP Kit (Wako) and quantified using the Quant-iT dsDNA Assay Kit (Invitrogen).
Statistical analysis
All statistical analyses were conducted using JMP Pro V.12.2.0. Non-parametric Mann-Whitney U test was used for comparisons between two groups and Steel test was used for comparisons between more than three groups. P values <0.05 were considered statistically significant. Correlations between clinical parameters and ISG-inducing activity or IFN-I level were determined using Spearman’s rank correlation coefficient (ρ).
Results
Elevated IFN-I bioactivity in SLE serum
In the Osaka University Hospital database, we searched for patients with SLE who satisfied the ACR revised criteria for classification of SLE.36 Sixty-four patients with SLE were enrolled in this study (see online supplementary figure S1). The clinical characteristics of patients and HCs are summarised in table 1. For comparison, 24 patients with SSc37 and 11 patients with SjS38 were also enrolled (see online supplementary table S1). To measure the bioactivity of IFN-I in serum, we used HEK-Blue IFN-α/β, which produces SEAP in response to IFN-I (see online supplementary figure S2A,B). IFN-I bioactivity was significantly higher in SLE sera than in sera from HC or patients with other connective tissue diseases (figure 1A).

High IFN-I levels and ISG-inducing activities of SLE serum. (A) IFN-I bioactivities in sera. IFN-I bioactivities in SLE (n=54), SjS (n=11), SSc (n=24) and HC (n=24) were measured using HEK-Blue IFN-α/β reporter cells. (B) ISG-inducing activities in sera. Levels of ISG-inducing activity in SLE (n=60), SjS (n=11), SSc (n=24) and HC (n=24) were measured using THP1-ISG reporter cells. (C) ISG-inducing activity of SLE sera with IFN-I signalling blockade. THP1-ISG reporter cells were stimulated with sera from SLE (n=48) and HC (n=7) in the absence (IFN blockade −) or presence (IFN blockade +) of antibody cocktail (anti-IFN-α, anti-IFN-β and anti-IFNAR2). (D) IFN-I production in PBMCs induced by SLE sera. Recombinant human IFN-α(rhIFNα), SLE sera (n=12) or HC sera (n=5) were incubated for 24 hours with or without PBMCs from healthy donors. Levels of IFN-I in the culture supernatants were measured using HEK-Blue IFN-α/β reporter cells. PBMC-derived IFN-I levels were calculated by subtracting the IFN-I levels in PBMC-absent medium from the IFN-I levels in PBMC-present medium. The box chart indicates the 75th percentile (upper), median (middle) and 25th percentile (lower) (A–C). Overall statistical differences between SLE and other groups were evaluated by Steel test (A,B). Overall differences between the groups were evaluated by Mann-Whitney U test (C,D). HC, healthy control; IFN-I, type I interferon; ISG, IFN-I-stimulated genes; PBMC, peripheral blood mononuclear cells; SjS, Sjögren syndrome; SLE, systemic lupus erythematosus; SSc, systemic sclerosis.
Characteristics of patients with SLE and HC
Induction of ISGs by SLE serum
Progression of SLE is associated with upregulation of IFN-I and ISGs.4–6 Using THP1-ISG cells, we investigated whether SLE serum could promote further IFN-I production. These cells secrete SEAP in response to various ISG-inducing stimuli, including IFN-I, 2’3’-cGAMP, cyclic-diAMP (c-diAMP) and lipopolysaccharide (see online supplementary figure S2C,D). ISG-inducing activity was considerably higher in SLE sera than in sera from HC, SjS or SSc (figure 1B). Although ISGs are upregulated in the PBMCs of SLE, SjS and SSc,41 42 only SLE sera had significant ISG-inducing activity.
IFN-I-independent ISG-inducing activity in SLE serum
We next examined the contribution of IFN-I in ISG induction by blocking IFN-I with antibodies against IFN-α, IFN-β and IFNAR2 (see online supplementary figure S3A). Although ISG induction was reduced by IFN-I signalling blockade, ISG-inducing activity still remained in SLE sera (figure 1C). To confirm this, we established IFNAR2-knockout THP1-ISG (THP1-ISG-KOIFNAR2) (see online supplementary figure S3B,C).40 Even in these cells, ISG-inducing activity was observed with SLE sera (see online supplementary figure S3D). We then evaluated the effect of SLE serum in human PBMCs. PBMCs from healthy donors did not produce additional IFN-I on stimulation with recombinant IFN-α/β (see online supplementary figure S3E). However, SLE sera induced significant IFN-I production in the PBMCs (figure 1D). Thus, ISG induction by SLE serum is driven by not only IFN-I but also other factors.
Association of ISG-inducing activity and TLR9
Loss of tolerance to nucleic acids and impaired DNA degradation are thought to be important in SLE pathogenesis.18 19 We purified and quantified extracellular DNA in sera and found that the amount of dsDNA was higher in SLE than in HC (figure 2A). Because TLR9 is a major dsDNA sensor,11 we investigated its involvement in SLE serum-mediated inflammation. THP1-ISG was unresponsive to agonists of TLR9 (figure 2B and online supplementary figure S4A), TLR7 and TLR8 (see online supplementary figure S4B). To assess the involvement of TLR9, we used HEK-Blue TLR9 (see online supplementary figure S4C,D). Some SLE sera activated HEK-Blue TLR9, but this activation did not correlate with the levels of IFN-I-independent ISG-inducing activity (figure 2C). Because TLR7 plays important roles in SLE pathogenesis,43 we examined the involvement of TLR7 and TLR8 (see online supplementary figure S4E,F). Although sera from several patients activated TLR7 and TLR8, the levels of activation did not correlate with IFN-I-independent ISG-inducing activity (see online supplementary figure S5A,B). Therefore, pathways other than TLR7, TLR8 or TLR9 mediated the ISG induction by SLE sera.

Elevated levels of dsDNA in SLE serum and involvement of TLR9. (A) dsDNA in sera. DNA was purified from SLE (n=64) and HC (n=18) sera and quantified. (B) THP1-ISG stimulated with TLR9 agonists. THP1-ISG cells were stimulated for 24 hours with STING agonist (5 µg/mL 2’3’-cGAMP), TLR9 agonist (10 µg/mL D35, 500 nM ODN2216, 10 µg/mL ODN2336, 10 µg/mL ODN2006, 10 µg/mL K3), control medium (Ctrl) or sera from SLE (n=64) and HC (n=17). (C) IFN-I-independent ISG-inducing activities and correlation with activation of TLR9. HEK-Blue TLR9 cells were stimulated with sera from SLE (n=22) and HC (n=20) for 24 hours. Levels of TLR9 pathway activation induced by sera from SLE (red circle) or HC (blue circle) are shown in the left panel. Correlation between TLR9 activation (y-axis) and ISG-inducing activity following IFN-I blockade (x-axis) is shown in the right panel. The box chart indicates the 75th percentile (upper), median (middle) and 25th percentile (lower) (A,B). Statistical analysis was performed by Mann-Whitney U test (A, B and left panel of C) and Hoeffding’s D statistic (right panel of C). cGAMP, cyclic GMP–AMP; dsDNA, double-stranded DNA; HC, healthy control; IFN-I, type I interferon; ISG, IFN-I-stimulated genes; SLE, systemic lupus erythematosus; STING, stimulator of interferon genes; TLR9, Toll-like receptor 9.
Induction of ISGs by SLE serum via the STING pathway
The cGAS–STING pathway is another dsDNA sensor.12 To evaluate its contribution in SLE serum-mediated inflammation, we established STING-knockout THP1-ISG (ISG-KOSTING) (figure 3A). When these cells were stimulated with SLE sera, most samples exhibited reduced ISG-inducing activity (figure 3B), suggesting that ISG induction was at least partially dependent on STING pathway. STING-dependent ISG-inducing activity correlated with the IFN-I-independent ISG-inducing activity of SLE sera (figure 3C). Thus, activation of the STING pathway may be responsible for high IFN-I levels in SLE.
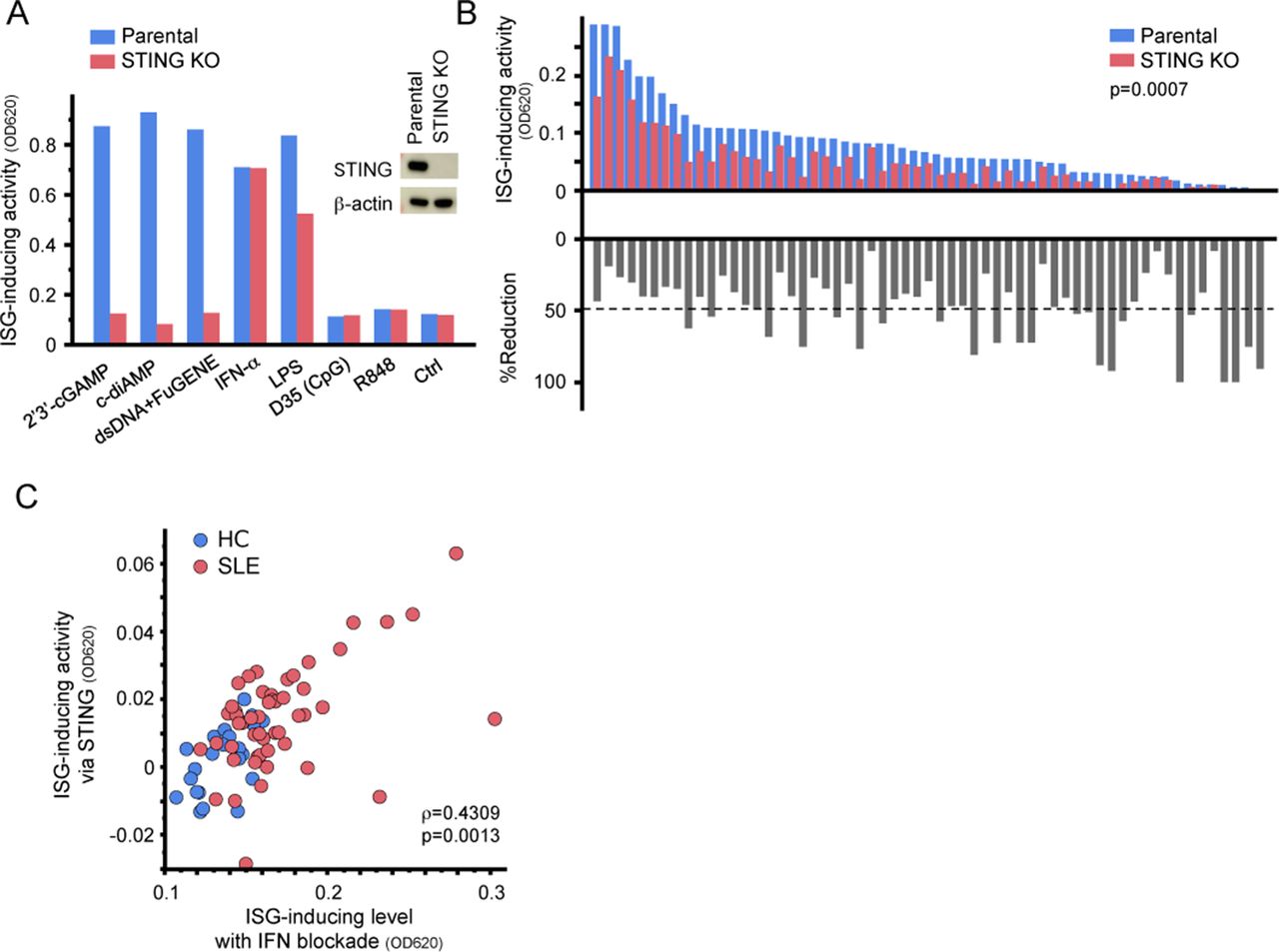
Serum-induced ISG expression via STING. (A) Establishment of STING-knockout reporter cells (ISG-KOSTING). Expression of STING was evaluated by western blotting. ISG-KOSTING cells did not respond to STING agonists (bar graph). Parental THP1-ISG (Parental) and ISG-KOSTING cells were stimulated with STING agonists (5 µg/mL 2’3’-cGAMP, 10 µg/mL c-diAMP), cGAS agonists (dsDNA+FuGENE), cytokine (1000 U/mL IFN-α), TLR agonists (500 ng/mL LPS, 10 µg/mL D35, or 10 µg/mL R848) or control medium (Ctrl) for 24 hours. Statistical analysis was performed by Mann-Whitney U test. (B) ISG-inducing activity in THP1-ISG-KOSTING. Parental THP1-ISG and THP1-ISG-KOSTING cells were stimulated with SLE sera (n=59) for 24 hours (upper bars). Each bar represents serum from an individual patient. %Reduction indicates the rate of reduction in reporter activity of THP1-ISG-KOSTING relative to that of parental THP1-ISG (lower bars). Dashed line indicates the mean of %reduction (C) Correlation between IFN-I-independent ISG-inducing activity and STING-dependent ISG-inducing activity. STING-dependent ISG-inducing activity was calculated by subtracting the absorbance of SEAP in THP1-ISG-KOSTING from that in parental THP1-ISG. (B). Statistical analysis was performed by Mann-Whitney U test (B) and non-parametric Spearman’s rank test (C). c-diAMP, cyclic-diAMP; cGAMP, cyclic GMP–AMP; cGAS, cyclic GMP–AMP synthase; dsDNA, double-stranded DNA; HC, healthy control; IFN-I, type I interferon; ISG, IFN-I-stimulated genes; LPS, lipopolysaccharide; SEAP, secreted embryonic alkaline phosphatase; SLE, systemic lupus erythematosus; STING, stimulator of interferon genes; TLR, Toll-like receptor.
Activation of the cGAS–STING pathway by AdMVs in SLE serum
The STING agonist 2’3’-cGAMP is synthesised by cGAS after dsDNA recognition.12 Hence, we investigate whether STING agonists were present in serum. Serum was analysed using triple quadrupole-type liquid Chromatography-Mass spectrometry (LCMS), which was capable of detecting 2’3’-cGAMP and c-diAMP at concentrations as low as 100 fmol/mL (see online supplementary figure S6A). Eight samples with STING-dependent ISG induction were analysed; however, neither 2’3’-cGAMP nor c-diAMP was detected (see online supplementary figure S6B). Consistent with this, ISG induction by SLE sera was not reduced even after cGAMP degradation with ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) (see online supplementary figure S6C).44 Thus, 2’3’-cGAMP and c-diAMP were not present in SLE serum.
Next, we examined the involvement of dsDNA in ISG induction. However, ISG-inducing activity was not diminished by DNase I treatment (figure 4A). We also investigated the involvement of ICs, but neither anti-FcγRI (CD64) antibody nor non-specific polyclonal IgG suppressed ISG induction by SLE sera (see online supplementary figure S7A,B). Hence, we examined the role of extracellular membrane vesicles in serum. To this end, we isolated AdMVs from serum (see online supplementary figure S8A–C). dsDNA was contained in AdMVs and protected from DNase I degradation (figure 4B,C). We then cultured PBMCs with the isolated AdMVs and measured IFN-I in the culture supernatant. AdMVs from SLE sera facilitated IFN-I production, whereas those from HC did not (figure 4D). In addition, AdMV-induced ISG expression in THP1-Dual, a THP1-ISG reporter with luciferase activity instead of SEAP production, was decreased by knockout of cGAS or STING (figure 4E and online supplementary figure S9A,B). These results indicate that AdMVs in SLE serum promote IFN-I production and ISG induction through the cGAS–STING pathway.
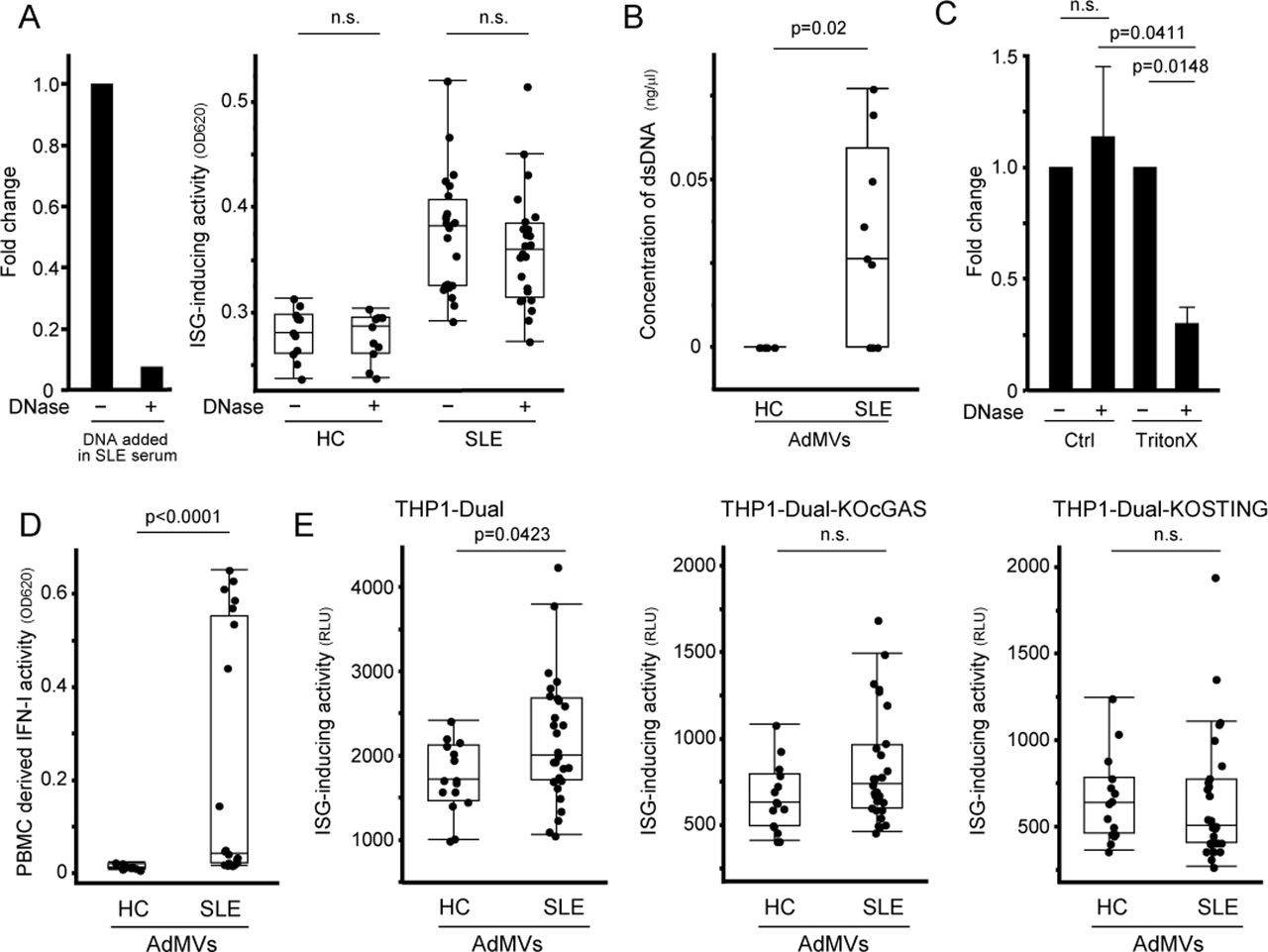
Induction of IFN-I production and ISG expression by apoptosis-derived membrane vesicles from SLE serum. (A) ISG-inducing activity of SLE sera after DNase I treatment. DNase I activity in serum was confirmed by degradation of DNA added in SLE serum (left). After sera from SLE (n=24) and HC (n=11) were pretreated with or without DNase I, THP1-ISG was stimulated with pretreated sera for 24 hours (right). (B) Concentration of dsDNA in AdMVs from sera. DNA was purified from AdMVs of SLE (n=9) and HC (n=6) and quantified. (C) Degradation of dsDNA in AdMVs from SLE (n=6) using DNase I with or without membrane-rupturing treatment. Membrane of AdMVs was permeabilised with Triton X-100 prior to DNase I treatment. (D) Induction of IFN-I production in PBMCs by AdMVs from SLE sera. AdMVs isolated from SLE (n=21) and HC (n=11) sera were cocultured with PBMCs from healthy donors for 24 hours. IFN-I bioactivity in the culture supernatants was measured using HEK-Blue IFN-α/β. (E) cGAS and STING-dependent ISG induction by AdMVs. THP1-Dual, THP1-Dual-KOcGAS and Dual-KOSTING cells were cultured with AdMVs from SLE (n=29) or HC (n=15) for 24 hours. ISG-inducing activity was evaluated by measuring luciferase activity in culture supernatants. The box chart indicates the 75th percentile (upper), median (middle) and 25th percentile (lower) (right panel of A, B, D, E). Values are mean±SEM (C). Statistical analysis was performed by Mann-Whitney U test (A, B, D, E) or Steel test (C). AdMV, apoptosis-derived membrane vesicles; cGAS, cyclic GMP–AMP synthase; dsDNA, double-stranded DNA; HC, healthy control; IFN-I, type I interferon; ISG, interferon-stimulated genes; PBMC, peripheral blood mononuclear cells; SLE, systemic lupus erythematosus; STING, stimulator of interferon genes.
Clinical features and ISG-inducing activity of SLE serum
Finally, we investigated the relationship between clinical features and the IFN-I bioactivity or ISG-inducing activity of SLE serum. As shown in figure 5A, the ISG-inducing activity of SLE sera correlated with the SLEDAI-2K score.39 Likewise, the IFN-I bioactivity, STING-dependent ISG-inducing activity and IFN-I-independent ISG-inducing activity also individually correlated with SLEDAI-2K (see online supplementary figure S10A). Patients with high ISG-inducing activity manifested lymphocytopaenia, low serum C4 levels and high anti-dsDNA antibody titres (figure 5B–D and online supplementary figure S10B). The presence of lupus nephritis did not affect the levels of IFN-I or ISG-inducing activity (figure 5E and online supplementary figure S10C). These results suggested that the ISG-inducing activity of serum measured by reporter cells could be a useful biomarker for assessing SLE disease activity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationships between ISG-inducing activity of SLE serum and clinical features of SLE. (A) Correlation between ISG-inducing activity and SLE disease activity, assessed according to SLEDAI-2K. (B–D) Association of ISG-inducing activity and clinical features of SLE. Patients with high levels of ISG-inducing activity manifested lymphocytopaenia (B), low serum C4 levels (C) and high anti-dsDNA antibody titres (D). All clinical data were obtained from the medical records of Osaka University Hospital. (E) ISG-inducing activity and lupus nephritis. Patients with SLE were divided into two groups according to the presence (n=35) or absence (n=29) of lupus nephritis. ISG-inducing activity of SLE sera (n=64) was evaluated using THP1-ISG cells (A–E). The box chart indicates the 75th percentile (upper), median (middle) and 25th percentile (lower) (E). Statistical analyses were performed by non-parametric Spearman’s rank test (A–D) or Mann-Whitney U test (E). dsDNA, double-stranded DNA; IFN-I, type I interferon; ISG, IFN-I-stimulated genes; SLE, systemic lupus erythematosus.
Discussion
In this study, we showed that SLE serum induced ISGs, in part, through cGAS–STING pathway. In addition, we found that dsDNA was present in SLE serum, and that AdMVs in SLE serum facilitated ISG expression via the cGAS–STING pathway. Using a cell-based reporter system, we demonstrated that the levels of IFN-I bioactivity and ISG-inducing activity were associated with SLE disease activity. Together, our results elucidate a possible pathological mechanism of SLE in which AdMVs drive IFN-I production through the cGAS–STING pathway (see online supplementary figure S11).
TLRs are major receptors in innate immunity that recognise nucleic acids. Endosomal TLRs are important in pathogenic overexpression of IFN-I and ISGs in SLE.15 17 22 43 45 46 However, the identification of cGAS and STING26–28 implicated their roles in the onset of SLE. In murine models, the role of cGAS–STING seems to be controversial. For instance, Sting−/− MRL/Faslpr mice exhibit more severe disease phenotype,47 while deficiency of cGAS or STING in DNaseII−/− mice results in reduced lupus-like symptoms.30 32 35 In humans, constitutively active mutations of STING are associated with SLE-like diseases.33 34 Although these studies suggest that the cGAS–STING pathway contributes to lupus pathophysiology, it remains unclear whether cell intrinsic or extrinsic factors activate this pathway. Recent studies reported that cGAMP is present in SLE-derived PBMCs,35 and that cGAMP is released into the extracellular space upon stimulation.48–50 We studied this phenomenon using ultra-high-sensitivity LCMS, but detected neither 2’3’-cGAMP nor c-diAMP in the SLE sera. However these experiments were limited by the small sample size. Here, we showed that the amount of dsDNA in SLE sera was high. SLE serum-induced ISG expression was mediated by cGAS and STING, suggesting that cell-extrinsic dsDNA activated the cGAS–STING pathway. Consistent with our findings, oxidised mitochondrial DNA released by NETosis can drive the cGAS–STING pathway.51 52 Our study did not address the characteristics of dsDNA in serum, such as fragment size, physicochemical properties, methylation status and histone modification. Therefore, further investigation of these features is necessary.
One possible delivery mechanism for extracellular dsDNA into the cytosol is engulfment of ICs.53 We assessed the contribution of ICs in SLE serum-mediated ISG induction by blocking FcγR. However, blocking engulfment of ICs did not affect ISG induction, suggesting that ICs did not mediate ISG-inducing activity in our experimental setting.
We observed that AdMVs isolated from SLE serum contain dsDNA and induce ISG expression. Nucleic acids present in extracellular vesicles are protected from enzymatic degradation.54 In accordance with this, dsDNA in AdMVs was resistant to DNase I and SLE serum-mediated ISG induction was unchanged by DNase I treatment. Consistent with our findings, SLE-derived AdMVs can activate immune cells, resulting in enhanced IFN-I secretion.55–57 In most studies, AdMVs are harvested from culture supernatant of PBMCs in which apoptosis has been artificially induced by ultraviolet radiation. By contrast, using a cell-based reporter system, we directly showed that AdMVs in SLE sera induced IFN-I production. Additionally, we demonstrated that AdMV-mediated ISG induction was cGAS-dependent and STING-dependent. In regard to the difference of AdMVs from SLE and those from HC, there are several possibilities: increased amount of AdMVs in SLE serum, increased amount of dsDNA in AdMVs, unique property of dsDNA in AdMVs, the nature of the membrane of AdMVs which may allow efficient engulfment by scavenger cells and so on. In our results, the amount of dsDNA in AdMVs was higher in SLE than that in HC. However, we could not address other possibilities. Further studies are needed to clarify the characteristics of AdMVs and their association with clinical manifestations.
Human TLR9 is highly expressed in certain immune cells, but at low levels in monocytes, macrophages and non-haematopoietic cells.58 59 Nonetheless, high ISG expression has been observed in these cell types in patients with SLE. Several studies showed that TLR9 expression is downregulated in the PBMCs of SLE.16 60 Additionally, the TLR9 response of plasmacytoid dendritic cells is transient and decreases with continuous exposure to ligands,24 25 suggesting that TLR9-independent mediators are important for chronic and durable IFN-I production. The THP1 cells we used were unresponsive to agonists of TLR7, TLR8 and TLR9. Therefore, THP1 is suitable for evaluating the involvement of cytosolic nucleic acid sensors other than TLRs. Various cytosolic DNA sensors (eg, TLR9, cGAS, AIM2 and others) recognise dsDNA,11 12 but functionally dominant sensors may depend on the cell type and inflammatory condition. In cells expressing low levels of TLR9, the cGAS–STING pathway may be the major sensor of extracellular DNA that ultimately leads to sustained IFN-I production in SLE.
In some patients with SLE, the dependence of IFN-I production on the cGAS–STING pathway was partial, suggesting the involvement of other factors. Cytosolic RNA can activate IFN-I production.11 Some SLE sera activated reporter cells for TLR7 and TLR8, suggesting that RNA may also be present in SLE serum. It is possible that RNA-sensing pathways might be involved in the SLE serum-induced IFN-I production. Therefore, further investigation of the roles of RNA sensors in SLE is warranted.
In terms of clinical indicators, we demonstrated that both IFN-I and ISG-inducing activity were higher in sera of SLE than other autoimmune diseases. The levels of IFN-I and ISG induction correlated with SLE disease activity. Stratification of patients with SLE based on molecular profiles is important because the clinical symptoms and gene expression profiles are heterogeneous.1 2 8 The expression levels of ISGs are thought to be useful biomarkers for SLE stratification. Accordingly, treatments that target IFN-I have been shown to be effective in patients with high ISG expression.9 10 However, the current method for evaluating ISG expression is still complicated,9 10 41 and the sensitivity of ELISA is insufficient.6 24 Because our cell-based reporter system enables easy and highly sensitive evaluation of both IFN-I and ISG induction, it may be useful for stratifying patients with SLE.
It should be noted that the number of samples in this pilot study might be insufficient to definitively determine the clinical significance of serum ISG-inducing activity. Also, the possibility of sampling bias cannot be excluded. Moreover, several issues still require clarification: whether the ISG-inducing activity of serum depends on the clinical features and whether measurement of ISG-inducing activity is useful for evaluating or predicting clinical progression and efficacy of treatment. Multicentre and prospective studies are needed to answer these questions.
Collectively, we have shown that SLE serum significantly induces ISG expression and IFN-I production. In mechanistic terms, AdMVs facilitate the IFN-I-producing cascade in a cGAS–STING-dependent manner. Accordingly, we propose the following model of SLE pathophysiology. First, defective clearance of apoptotic cells produces AdMVs containing dsDNA, which in turn induces IFN-I production via the cGAS–STING pathway. Next, IFN-I activates immune responses that lead to tissue damage in various organs, resulting in further generation of AdMVs, triggering a positive-feedback loop of IFN-I production and further tissue damage (see online supplementary figure S11). In this context, it may be promising to target the cGAS–STING pathway for SLE treatment, and our cell-based reporter system may be useful for stratifying patients with SLE with high ISG-inducing activity.
Acknowledgments
The authors appreciate the patients and healthy volunteers who participated in this research. We also thank the members of the Department of Clinical Immunology for collection of blood samples. We are grateful to Dr Hiroyuki Miyoshi (Keio University, Japan), the RIKEN BioResource Center (RIKEN BRC) and Dr Feng Zhang (MIT, USA) for providing the materials.
References
Footnotes
YK and JHP contributed equally.
Handling editor Josef S Smolen
Contributors HT and YKa designed the project. YKa and JP performed most experiments. YKa analysed the data. HT, YKa and JP wrote the manuscript. SA and WA performed the MS analysis. HK assisted with the experiments. SK, YH and YKi participated in the discussions. MNi, TH, YS and MNa collected clinical samples and provided critical suggestions. MU provided critical collaborative suggestions regarding the MS analysis. AK oversaw the study.
Funding This study was supported in part by a research grant from JSPS KAKENHI (15K09550, to HT); a Core Research for Evolutionary Science and Technology (CREST) grant from the Japan Science and Technology Agency (JST) (JPMJCR16G2, to HT and MU); the Naito Foundation (to HT); the Center of Innovation Program (COI-STREAM) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (to AK); Japan Agency for Medical Research and Development (AMED)-CREST grant (15652237, to AK); and MEXT Grant-in-Aid (14525051, to AK).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Enrolment of all participants was approved by the institutional review boards of Osaka University. Informed consent was obtained from all subjects in accordance with the Declaration of Helsinki and with approval of the ethical review board of Osaka University (no 12456-3 and 11122-4).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available from the corresponding author upon request.