Article Text

Abstract
Background Despite growing interest, there is no guidance or consensus on how to conduct clinical trials and observational studies in populations at risk of rheumatoid arthritis (RA).
Methods An European League Against Rheumatism (EULAR) task force formulated four research questions to be addressed by systematic literature review (SLR). The SLR results informed consensus statements. One overarching principle, 10 points to consider (PTC) and a research agenda were proposed. Task force members rated their level of agreement (1–10) for each PTC.
Results Epidemiological and demographic characteristics should be measured in all clinical trials and studies in at-risk individuals. Different at-risk populations, identified according to clinical presentation, were defined: asymptomatic, musculoskeletal symptoms without arthritis and early clinical arthritis. Study end-points should include the development of subclinical inflammation on imaging, clinical arthritis, RA and subsequent achievement of arthritis remission. Risk factors should be assessed at baseline and re-evaluated where appropriate; they include genetic markers and autoantibody profiling and additionally clinical symptoms and subclinical inflammation on imaging in those with symptoms and/or clinical arthritis. Trials should address the effect of the intervention on risk factors, as well as progression to clinical arthritis or RA. In patients with early clinical arthritis, pharmacological intervention has the potential to prevent RA development. Participants’ knowledge of their RA risk may inform their decision to participate; information should be provided using an individually tailored approach.
Conclusion These consensus statements provide data-driven guidance for rheumatologists, health professionals and investigators conducting clinical trials and observational studies in individuals at risk of RA.
- arthritis
- rheumatoid
- autoimmunity
- arthritis
- autoantibodies
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
It is now clear that the onset of rheumatoid arthritis (RA) is preceded by a complex preclinical phase.1 While the early arthritis paradigm (ie, early identification and treatment) has revolutionised the outlook of RA, interventions targeting the preclinical phase may unleash an even greater therapeutic leap. In the preclinical phase, ‘at-risk’ individuals, many of whom have genetic or environmental predispositions, develop autoantibodies and/or symptoms and eventually progress to clinical arthritis and classifiable RA.2 Over the last decade, longitudinal observational studies of prospective at-risk cohorts have identified risk factors and biomarkers, which have enabled a better understanding of RA pathobiology and also prediction of the onset and timing of clinical arthritis.3–5 In this way, symptomatic at-risk populations may now be risk stratified for future RA development.6 7 Building on this work, clinical trials investigating therapeutic intervention in at-risk individuals with the aim of RA prevention have been conducted.8 9 Several more are now either underway or in preparation; all are targeting at-risk populations with the aim of RA prevention. While sharing a common goal, these trials are strikingly heterogeneous; different at-risk populations have been included with different eligibility criteria, biomarkers, interventions and outcomes. The heterogeneity is a natural consequence of the infancy of the field but may present unwelcome challenges in interpreting the relevance and validity of the findings. As this field grows exponentially, it is critical that all future efforts are optimally aligned; at-risk individuals are difficult to identify, recruit and monitor but provide invaluable opportunity for insights into the pathobiology of RA and new avenues for prevention that must be maximised.
The current EULAR task force was convened with the goal of providing data-driven guidance and consensus for use by current and future investigators in this important area of rheumatology research.
Methods
An international multidisciplinary task force was convened with the aim of defining points to consider for conducting clinical trials and studies in individuals at risk of RA (co-convened by KM and PE). The task force included 13 academic rheumatologists from Europe and North America with specific expertise in this area. There were two project methodologists/epidemiologists (DA and AK). The task force also included one health professional (HP), two rheumatologists from the EMerging EULAR NETwork (EMEUNET) and two patient representatives from the people with arthritis / rheumatism across Europe (PARE) network of patient research partners. In developing the points to consider, the task force followed the most recent EULAR standardised operating procedures for the development of recommendations.10 The project was fully approved by the EULAR executive committee.
At the first meeting (October 2019 in Amsterdam, the Netherlands), the task force discussed the background and focus of the project and defined the objectives. Four key questions to be addressed by systematic literature reviews (SLRs) were then prioritised by a group voting process, supervised by the methodologists. The SLR was performed by the fellow and co-convenor (KM) and the allied health professional (AHP) (HJS) with support from one of the EMEUNET members (DAR), a librarian (Joel Kerry) and a research fellow from Leeds (Andrea Di Matteo). Based on the findings of the SLRs, a draft of the points to consider and research agenda was prepared by the fellow (KM), AHP (HJS), the methodologists (DA and AK) and the convenor (PE).
At the second meeting (held in April 2020, by video conference due to the COVID-19 pandemic), the SLR results and the draft of the points to consider and research agenda were presented to the task force. Following group discussions, during and after the meeting, the task force agreed on the final consensus statements which encompassed 1 overarching principle, 10 points to consider and 1 research agenda. Task force members were then asked to anonymously rate the overarching principle and points to consider on a scale of 0 (absolutely disagree) to 10 (absolutely agree) to assess the level of agreement (LoA). The research agenda was extensively discussed between members and consensus was achieved on the points to be included. Comments from external industry stakeholders (Marie Brazil, Francesco De Leonardis and Jens Gammeltoft Gerwien) on the final consensus statements were proactively elicited and further considered during the writing of the manuscript.
Results
Systematic literature review
The task force agreed on the following four questions to be addressed by the SLR:
In clinical studies involving individuals at risk of RA, which populations should be included and what study endpoints should be used?
In individuals at risk of RA, is there a core set of risk factors and how frequently should they be measured?
In individuals at risk of RA, does risk-factor-driven intervention alter risk of progression?
Is there a benefit in informing individuals at risk of RA about their risk of developing RA and offering preventive treatment?
It was acknowledged that questions 3 and 4 were focused on potential interventions. The task force felt these questions would inform the design of clinical trials and studies in this area and would be important to include; question 3 would inform which potential interventions should be selected by investigators, while question 4 would inform recruitment strategies, communication approaches and feasibility of future studies.
To address these questions, four separate literature searches were conducted (see online supplemental materials for details). For each search, the relevant keywords were used in Medline, Embase, Pubmed and Central databases. Abstracts from January 2018 onwards were included. Meta-analyses were included, but all other reviews and study protocols were excluded. Manually searched articles either from the references of selected manuscripts or identified by task force members were also included.
Supplemental material
The task force agreed on 1 overarching principle, 10 points to consider (table 1) and 1 research agenda. When deciding overarching principles, discussion focused on the at-risk populations being recruited, data collection, study design and outcomes. Consideration was given to principles included in the 10 points to consider, to ensure specific guidance included in these points was not repeated. It was agreed that only one overarching principle should be put forward and this should specify key features, which should be collected from all at-risk populations.
Overarching principle and points to consider for conducting clinical trials and observational studies in individuals at risk of RA
Industry stakeholders received the initial draft of the manuscript and made comments and edits mainly around text wording, communication of content and structure. For example, statements were clarified with explanatory text where needed. The industry stakeholders helped ensuring that the manuscript would be relevant and accessible to potential industry partners who may be involved in future clinical trials and studies in this area. Industry bias was avoided as representatives were chosen to provide personal views based on their individual expertise and experience only (two in clinical research and trials and one in basic and translational science).
Overarching principle
All clinical trials and observational studies in individuals at risk of RA should include the epidemiological and demographic characteristics of the at-risk population being studied (LoA 10)
The task force recognised the different populations of at-risk individuals currently being studied in prospective cohorts internationally. While individual studies may prioritise the investigation of specific risk factors or a specific intervention in these populations, the task force agreed that certain population characteristics should invariably be measured. These are the core epidemiological and demographic characteristics of age, sex, body mass index (BMI), ethnicity, smoking status and family history of RA. Recording these core characteristics enables direct comparison and where possible integration of datasets from multiple cohorts.
Points to consider
1. For clinical trials or observational studies, individuals at risk of RA should be identified according to their clinical presentation. Within each clinical presentation, subpopulations should be identified based on the presence of specific risk factors (LoA 9.75)
Several different populations of individuals at risk of RA are being included in prospective clinical studies and interventional trials. The population differences largely reflect the available infrastructure, local populations and research interests of the various centres involved. Individuals usually present to healthcare professionals because of their clinical symptoms and signs, and the evolution of clinical features also reflects the natural history of RA development. Therefore, the task force felt it appropriate for at-risk populations to be categorised according to their clinical presentation. The task force decided against categorising at-risk populations based on just ‘symptoms’ and ‘signs’, as clinical signs (eg, joint tenderness) may be present in the absence of clinical synovitis. The term ‘arthralgia’ was avoided in categorisation as not all at-risk individuals with musculoskeletal (MSK) symptoms have arthralgia, with some presenting with non-specific symptoms instead. The task force proposed three broad categories, which underpin disease progression: asymptomatic, MSK symptoms without clinical arthritis and early clinical arthritis.
Asymptomatic at-risk individuals
Asymptomatic at-risk individuals are typically identified through either family relationships or population screening for the presence of informative autoantibodies.11–13 With these approaches, asymptomatic individuals are presumed to exhibit genetic or environmental risk factors without clinical symptoms or signs of arthritis. The influence of genetic and environmental risk factors in asymptomatic individuals who exhibit autoantibodies has not been well characterised across prospective cohorts. The first-degree relatives (FDRs) of people with RA are a population with genetic risk who may be feasibly identified from the general population (via the affected RA proband). FDRs are currently being studied in multiple research cohorts both to understand the pathobiology of RA and to investigate the influence of specific risk factors on disease progression in this population.14–19 Considering risk factors in FDRs, serum autoantibodies and other serum biomarkers have been the best characterised. Serum anti-citrullinated protein antibodies (ACPA) are enriched in FDRs20 and associated with arthritis development.17 Multiple serum cytokines and chemokines are associated with ACPA and disease progression in FDRs.21 22 Conversely, omega-3 fatty acid levels appear to have an inverse relationship with anti-cyclic citrullinated protein (anti-CCP) antibodies in those with genetic risk.23 24
In addition to FDRs from the general population, specific geographical populations also carry a heightened genetic risk of RA. Indigenous North Americans (INA), also referred to in the research literature as Indigenous Peoples, North American Natives, First Nations, First Nations Peoples, North American Indians, Aboriginals or Aboriginal peoples, have been the best characterised.25 26 Many of these populations exhibit high RA prevalence rates of predominantly seropositive, severe disease,27 familial clustering of cases19 and unfavourable disease outcomes. Although likely a significant factor, the increased risk may not be solely due to genetics, environmental factors, access to appropriate rheumatology care or a combination of these factors are also likely to be important. Studies of genetic risk in American Indians of Alaska and First Nations Peoples of Central Canada have shown that the shared epitope (SE) encoding allele HLA-DRB1*1402, which is almost unique to Indigenous Peoples, is a particularly important risk factor.28 Non-human leukocyte antigen (HLA) genes in these populations also appear to play a role.29
Longitudinal studies in FDRs of indigenous populations have demonstrated a high prevalence of serum ACPA (~10%) and rheumatoid factor (RF) (~15%). Although associated with arthritis development, ACPA levels fluctuate over time and not uncommonly revert to a seronegative state.30 Also, ACPA IgG variable domain glycosylation is a strong predictor of future RA development, in such populations.31
Overall, the task force agreed that FDRs, individuals who screen positive for ACPA and genetically predisposed indigenous populations are important asymptomatic at-risk populations that should be studied. Within these populations, serum autoantibodies and other serum biomarkers enable identification of important subpopulations.
At-risk individuals with MSK symptoms without clinical arthritis
Several different symptomatic at-risk populations without clinical arthritis are being studied. These include ACPA-positive individuals with MSK symptoms,7 32 seropositive (ACPA and/or RF) individuals with arthralgia33 and individuals with clinically suspect arthralgia (CSA).34 35
In these individuals, subpopulations may be defined based on serum autoantibodies, serum biomarkers, clinical symptoms and subclinical inflammation on imaging. In ACPA-positive individuals with MSK symptoms, a high anti-CCP level and the presence of RF are strongly associated with arthritis development.7 A high ACPA level is also associated with disease progression in ACPA-positive individuals without arthritis, but less well-defined clinical symptoms.36 37 In patients with seropositive arthralgia, the presence of anti-CCP antibodies, a high level of anti-CCP antibodies, the extent of the ACPA repertoire and dual positivity to anti-CCP and RF are all associated with arthritis development.6 33 38 39 ACPA and RF positivity are also associated with arthritis development in individuals with CSA.35 40 Anti-carbamylated antibodies have also been shown to be associated with arthritis development in patients with seropositive arthralgia.41 Serum and cellular biomarkers (T cell subsets) have predictive value in ACPA-positive individuals with MSK symptoms42 and seropositive arthralgia.43 44 The presence of certain clinical symptoms such as small joint tenderness and early morning stiffness are relevant for risk stratification in ACPA-positive individuals with MSK symptoms and patients with seropositive arthralgia.6 7 In patients with CSA, difficulty making a fist and a positive ‘squeeze test’ are associated with subclinical inflammation on MRI particularly tenosynovitis45 46 and is predictive of arthritis development.45 A set of clinical features in individuals at risk of RA were defined by a recent EULAR task force.34
In ACPA-positive individuals with MSK symptoms, patients with seropositive arthralgia and patients with CSA, subclinical inflammation on imaging has been characterised on ultrasound (US) and MRI. Intra-articular inflammation (grey scale and power Doppler signal) is the most relevant on US,47 48 whereas tenosynovitis is the most specific and predictive feature for arthritis development on MRI.49 50
The task force agreed that serum autoantibodies (especially ACPA), serum and cellular biomarkers, clinical features and subclinical inflammation on imaging should all be used to characterise subpopulations in symptomatic at-risk individuals without clinical arthritis.
At-risk individuals with early clinical arthritis
Two important populations with early clinical arthritis who are at risk of progression to RA are patients with undifferentiated arthritis (UA) and palindromic rheumatism (PR). Both have been studied extensively in prospective cohorts. For both conditions, clinically relevant subpopulations may be defined based on the presence of serum autoantibodies,51–54 serum biomarkers, clinical features54 and subclinical inflammation on imaging.55–57 In both PR and UA, these factors may be used for risk stratification (discussed below).
2. In longitudinal studies of individuals at risk of RA, the development of clinically evident arthritis, or progression to RA (according to 2010 ACR/EULAR Rheumatoid Arthritis Criteria), should be considered as study end-points (LoA 9.85)
Most longitudinal studies in at-risk populations have been performed in cohorts of individuals who have MSK symptoms without clinical arthritis (as described in point 1). The development of clinically evident arthritis is the most frequently studied primary end-point in longitudinal studies in ACPA-positive individuals with MSK symptoms, patients with seropositive arthralgia and patients with CSA. In many cases, individuals who develop clinical arthritis will also meet ACR/EULAR classification criteria for RA.58 This criterion is often included as a separate secondary end-point in studies. Investigators of a large Mexican longitudinal cohort study of relatives of RA probands (including FDRs) defined the development of clinical inflammatory arthritis (IA) as the primary end-point17 as have others.59 Longitudinal studies of INA have used development of RA as the primary end-point.30 60
Longitudinal studies of at-risk populations with clinical arthritis (ie, UA and PR) have specified the development of RA (either 2010 ACR/EULAR Criteria or previously accepted criteria) as the primary end-point. For PR, all such longitudinal studies have used the 1987 (or older) criteria due to age of the studies.51–53 61–65 In these studies, the primary end-point represents a clear transition from a relapsing–remitting phenotype to a persistent arthritis. In studies of patients with UA, the population is described as having undifferentiated or unclassified arthritis or early IA not meeting classification criteria for RA.66–76 In each case, the definition of UA is based on having clinical arthritis but not meeting the specified classification criteria for RA. Therefore, the RA classification criteria used are critical to the interpretation of the findings. The task force acknowledged that many of the patients with UA included in studies, which have not used the most recent classification criteria, are likely to have met the updated 2010 criteria, which is more sensitive for early disease.
3. The development of subclinical inflammation on US and/or MRI should also be considered as an end-point in at-risk populations without subclinical or clinical disease (LoA 8.65)
In at-risk populations without clinical or subclinical joint disease, the development of pathological subclinical inflammation on imaging is a significant step as it represents a transition from systemic autoimmunity to local articular inflammation, and is associated with imminent clinical arthritis. To reflect this, some longitudinal cohort studies stipulate the development of imaging-detected synovitis as the primary study end-point.77 Subclinical synovitis on US also influences clinical decision-making in at-risk individuals with MSK symptoms.78 The task force, therefore, felt it appropriate that the development of subclinical inflammation on imaging should be considered as an end-point distinct from the development of clinical arthritis (see table 2 for definitions of arthritis). The most appropriate imaging modality and protocol to use for detection of subclinical inflammation in at-risk populations are a subject for future research and were beyond the scope of the current task force.
Definitions of arthritis
4. In at-risk populations with clinical arthritis (ie, patients with PR and UA), interventional studies should include disease remission (on/off therapy) as an end-point (LoA 9.55)
Several interventional studies have been performed in patients with UA. Some have specified disease remission as the primary study end-point.68 79 80 In addition, others have included disease remission as a secondary end-point,81–84 with the primary end-point instead being the development of classifiable RA. Of those studies stipulating disease remission as an end-point, the majority tested short-term induction therapy, that is, the ability to achieve drug-free remission (DFR). In the PRObable rheumatoid arthritis: Methotrexate vs Placebo Treatment (PROMPT) trial, van Dongen et al investigated 12 months of induction therapy with methotrexate (MTX) in patients with UA and stipulated DFR at 30-month follow-up as a study end-point. There was no significant difference in DFR rates in the MTX and placebo arms.82 However, in a subanalysis of high-risk patients only (Leiden prediction score ≥8), DFR at 30 months was achieved in 36% of the MTX arm compared with 0% in the placebo arm (p=0.027), although in a total of only 22 patients.81 In the Stop Arthritis Very Early (SAVE) trial, the primary end-point was disease remission at 12 weeks and 15 weeks after a single intramuscular injection of methylprednisolone or placebo. DFR was achieved by 32/198 (16.2%) of the treatment group and 33/185 (17.8%) of the placebo group (p=0.68).79 In a recent observational study, data were used from the Induction therapy with MTX and prednisolone in Rheumatoid Or Very Early arthritic Disease (IMPROVED) study to specifically investigate predictors of DFR at 1 year in a subgroup of patients with UA who achieved remission and tapered all therapy at 8 months, according to a predefined protocol.80 In a recent small study of infliximab (IFX; given at week 0, week 2, week 6, week 14 and week 22) versus placebo in patients with ACPA-positive UA, DFR (according to DAS28 CRP) at 1 year was observed in 50% of the IFX group versus 21.4% of the placebo group.83
In contrast to the above-mentioned studies, two trials have used disease remission while still on therapy as an end-point.68 84 The first was a trial of IFX in patients with poor-prognosis UA who relapsed after a single corticosteroid injection, and the second a study of MTX compared with placebo. There was no difference in remission rates in the IFX study, whereas in the more recent MTX study, the proportion of patients who achieved Boolean remission after 1 year was greater in the MTX group compared with the placebo group.84
In a recent interventional study performed in a PR cohort, a predefined disease modifying anti-rheuamtic drug (DMARD) escalation protocol was used to bring flares under control with the aim of achieving disease remission.85 Complete or partial remission was achieved in 76/106 (82.6%) of patients, while 16.3% of patients were able to discontinue medications and achieve DFR. Disease remission (defined as the absence of flares for ≥1 month) on therapy was also the primary end-point in a study of rituximab (RTX) in PR.86 All of the 33 patients with seropositive PR in this study eventually achieved remission, although some required four cycles of RTX to do so. Neither of these studies were controlled trials.
5. In clinical trials or observational studies of individuals at risk of RA, risk factors should be assessed in a population-specific manner. Risk factors should include, or be a composite of, core and emerging risk factors (LoA 9.7)
Risk factors for the development of RA have been investigated using both large retrospective case–control studies and prospective cohort studies in predefined at-risk populations. The advantage of the former is the availability of large datasets from national and international registries, which allow the influence of specific genetic or environmental risk factors in the background population to be accurately quantified. However, to investigate the influence of specific risk factors in at-risk populations (as opposed to the general population), as well as the effects of risk factors that operate in a stage-specific manner, prospective cohort studies have proven most valuable.
Several different risk factors have been investigated in prospective cohort studies in well-defined at-risk populations. The task force agreed that risk factors are population specific, for example, symptom complexes and imaging are only relevant in at-risk populations who have symptoms. Therefore, risk factors should to be assessed according to the at-risk population, which is being included in a particular study or trial. Within each population, some risk factors have a strong evidence base, while others have a more limited evidence base. It was, therefore, felt that ‘core’ and ‘emerging’ risk factors may be defined based on the current evidence base. The task force agreed that core risk factors should, where feasible, always be assessed in a clinical trial or observational study (table 3).
Core and emerging risk factors for arthritis according to different at-risk populations
Asymptomatic at-risk individuals
Studies in large prospective cohorts of individuals with genetic risk factors for RA or identified in population screening have demonstrated that the additional presence of ACPA and other autoantibodies are significant risk factors for arthritis development. Anti-CCP-positive relatives (mainly FDRs) of RA probands are at much higher risk of developing RA compared with their seronegative counterparts (positive predictive value (PPV) of 64% for RA development at 5 years in CCP+/RF+ relatives).17 Similarly, 30% of anti-CCP+/RF+ relatives of INA developed RA after a median of 3-year follow-up.30 Before the availability of ACPA testing, a seminal study of over 2000 healthy INA monitored biannually for up to 19 years revealed a highly significant association between RF level and RA development (p<0.001, controlling for age and sex).60
Genotype also confers additional risk of ACPA and RA in FDRs of INA populations. The combination of HLA-DR SE and the non-SE allele DRB1*0901 is associated with ACPA and earlier age of RA onset in these FDRs.87 IgG ACPA glycosylation also appears to be strongly predictive of the future development of RA in INA FDRs.31
The core and emerging risk factors, which should be assessed in this population, are summarised in table 3.
At-risk individuals with MSK symptoms but without clinical arthritis
Several risk factors for the development of clinical arthritis and RA have been identified in ACPA-positive individuals with MSK symptoms, patients with seropositive arthralgia and patients with CSA. Autoantibodies (especially ACPA), clinical symptoms and imaging markers have the strongest evidence base.
The importance of ACPA status has been confirmed in various international cohorts, including a UK ACPA+ cohort with new non-specific MSK symptoms,7 a Dutch seropositive (CCP+/RF+) arthralgia cohort33 38 and a Dutch CSA cohort.40 The heightened risk conferred by high-tire ACPA is reflected in the highest weighting given to this risk factor in two clinical prediction rules for arthritis development.6 7 Anti-carbamylated protein (anti-CarP) antibodies also appear to confer additional risk of arthritis development in ACPA/RF+ at-risk individuals, independent of ACPA status.41 However, the level of risk does not appear to be as pronounced as that related to ACPA.
Certain clinical features are associated with an increased risk of arthritis development. Of these, prolonged early morning stiffness (EMS) duration is a cardinal symptom. Prolonged EMS duration is an important risk factor for arthritis in ACPA+ individuals with MSK symptoms (EMS >30 min)7 and patients with seropositive arthralgia (EMS >60 mins),6 and has been included in clinical prediction rules. EMS is also one of the components included in the agreed definition of CSA34 and is associated with arthritis development in unselected patients presenting with arthralgia.88 Joint tenderness (especially of the small joints) was also associated with arthritis development in ACPA+ individuals with MSK symptoms7 36 in two cohorts. Other symptom complexes were associated with progression to arthritis in seropositive arthralgia—duration of symptoms less than 1 year, intermittent symptoms and history of joint swelling.6 These symptoms were not prognostic in the UK ACPA+ cohort, perhaps because all individuals had new-onset symptoms and patients with PR were specifically excluded.
Abnormalities on high-sensitivity US and MRI (and positron emission tomography in one study89) also signify an increased risk for arthritis development in at-risk individuals. These abnormalities reflect the presence of intra-articular and extracapsular subclinical inflammation, in at-risk individuals who have symptoms without clinically evident arthritis. Several studies have identified the presence of subclinical US synovitis (power Doppler and grey scale) as a risk factor for arthritis development both at joint and patient level.47 48 90 91 Recent data suggest US tenosynovitis is associated with arthritis development in ACPA+ individuals with non-specific MSK symptoms and no subclinical synovitis on baseline US.77 92 In those with symptoms, the development of US synovitis appears to be a relatively late event, reflecting imminent clinical arthritis,93 especially as more joints become involved.94
MRI studies in ACPA+ individuals with MSK symptoms and patients with CSA have identified tenosynovitis as both the most prevalent abnormality and the strongest MRI risk factor for arthritis development.35 50 95 Tenosynovitis was the only MRI abnormality associated with arthritis progression at patient level in a study of 98 ACPA+ individuals with MSK symptoms (HR 4.02 (1.91–8.44), p=0.002).95 Similarly, tenosynovitis was the only MRI risk factor independently associated with arthritis development in a prospective study of 150 patients with CSA (HR 8.39 (3.38–20.81), p<0.001). There is also an increased prevalence of hand interosseous tendon inflammation in ACPA+ individuals with MSK symptoms.96 These studies highlight the importance of extracapsular inflammation (which MRI is particularly sensitive for) as a risk factor for disease progression.
Other clinical, cellular and serological risk factors for arthritis development have also been identified in this population, although current evidence for these factors is based largely on single unvalidated studies. Elevated BMI has been associated with arthritis development in some patients with seropositive arthralgia97 and ACPA+ at-risk individuals (some FDRs and some with MSK symptoms).98 Studies in the Dutch seropositive arthralgia cohort have shown that the number of peripheral blood B cell receptor clones,99 serum apolipoprotein A1100 and serum 14-3-3n levels101 are all associated with arthritis development. Loss of bone mineral density was associated with arthritis development in a prospective study in patients with CSA.102 Peripheral blood T cell subsets were also associated with arthritis development in ACPA+ at-risk individuals.42 The peripheral blood B cell signature103 and type I interferon signature104 have also been shown to be predictive of arthritis development in patients with seropositive arthralgia.
Early clinical arthritis
Clinical features, imaging findings and autoantibody profile are also important risk factors for the development of RA in patients classified as having UA. These data are largely based on prospective analyses of UA cohorts, with patients with early arthritis included on the basis of failure to meet now outdated versions of RA classification criteria. As such, it is likely that a significant proportion of these patients would now, based on current criteria,58 classify as early RA. Several risk prediction tools, combining clinical features, autoantibodies and/or imaging risk factors, have been proposed in UA cohorts.66 69–71 73 105–107 Many of the risk factors are the same as those described above for individuals with MSK symptoms without clinical arthritis; prolonged EMS duration,69 105 106 ACPA and/or RF,66 69 71 73 105 106 and power Doppler (PD) signal on US105 have all been shown to predict progression of UA to RA. However, elevated CRP/ESR,69 105 longer disease duration,73 105 greater number of swollen and/or tender joints69 73 and radiographic erosions69 70 are risk factors for progression specific to patients with early clinically apparent arthritis. Autoantibody profile (ACPA and RF) and flares involving the hands and wrists are also risk factors for RA development in patients with PR.51 52
Subclinical inflammation on US and MRI has been identified as a risk factor for RA development in several UA cohorts.108–113 The presence of US synovitis (gray-scale (GS) and/or PD) is predictive of progression from early UA to RA.110–112 114 However, tenosynovitis in the hands and feet113 on US and MRI also appears to be associated with disease progression.109 112 Of the studies investigating the role of autoantibody profiling in UA, ACPA status and level (high level most predictive) are the most consistently predictive of disease progression.67 72 74
There has been considerable interest in the putative role of mucosal inflammation and dysbiosis in initiating and driving disease progression in at-risk individuals.15 115–119 The potential importance of this was recognised by the task force. Although limited data were identified reporting these as specific risk factors for arthritis development,120 the task force felt that these factors should be considered in future studies and clinical trials.
Considering the available data, the task force proposed that specific core and emerging risk factors should be assessed in symptomatic at-risk populations, that is, at-risk individuals with symptoms (but no clinical synovitis) and individuals with early clinical arthritis (table 3).
6. Risk factors should be assessed at baseline and repeated assessment considered according to the specifics of the study population and intervention (LoA 9.75)
The majority of studies investigating risk factors in prospective at-risk cohorts have taken only a single baseline measurement of the risk factor(s). While this provides valuable information on the overall influence of risk factors on the development of arthritis, a single measurement cannot address whether and to what extent risk factors change over time, nor the relationship between different risk factors over time. For example, some risk factors may be more relevant in the asymptomatic at-risk phase, while others may become more relevant in symptomatic individuals when the onset of clinical arthritis is imminent.
In line with this, recent data suggest the prevalence and overall burden of subclinical joint inflammation on US imaging increases over sequential assessments prior to arthritis development in ACPA+ individuals with MSK symptoms.93 94 This suggests that US subclinical inflammation is most relevant in those with imminent arthritis. There is a paucity of longitudinal, repeated assessments of other risk factors, particularly the evolution of clinical symptoms or cellular/serological markers. Investigators may seek to understand the influence of interventions on risk factors (ie, surrogates of disease progression), as well as arthritis development. An understanding of the stability of risk factors will be critical to designing such studies. The task force felt that this was an area which should be prioritised in future clinical trials and longitudinal studies.
7. Clinical trials should evaluate the ability of a specific intervention to modify the risk factor itself, as well as the risk of progression to RA (LoA 9.65)
Clinical trials in individuals at risk of RA have primarily focused on the prevention of progression to clinical arthritis or RA. Given increasing evidence for the role of specific risk factors in driving disease progression (as detailed above), the task force felt that evaluation of the ability of interventions to modify an individual’s underlying risk factors should be prioritised in future study designs. This would be important for two reasons: first, to better understand the relative influence of specific risk factors on disease progression and second, it would represent an important step towards personalising different types of preventive intervention (eg, pharmacotherapies, lifestyle modifications or a combination) by understanding their suitability to target specific risk factors, which may be enriched in different at-risk populations.
Published studies suggest modification of risk factors in this way is feasible; improvement in RA-autoantibody levels has been reported in interventional trials in both patients with seropositive arthralgia and patients with UA.8 121 In the Abatacept study to determine the effectiveness in preventing the development of rheumatoid arthritis in patients with Undifferentiated inflammatory arthritis and to evaluate safety and tolerability (ADJUST) trial, induction therapy with abatacept (ABT) in patients with UA was associated with a reduction in anti-CCP level. MRI osteitis scores also improved with ABT therapy and this benefit was sustained 6 months after treatment withdrawal.121
8. In individuals at high risk of RA (eg, with early clinical synovitis), drug intervention should alter progression to RA or the outcome of RA therapy (LoA 9.5)
Clinical trials have shown that in patients with UA, drug intervention can reduce or delay progression to RA and improve the outcome of therapy.81 82 84 121–123 There is particularly strong evidence for a beneficial effect of MTX in UA.81 82 84 The Dutch PROMPT study showed a delay in development of RA when MTX and placebo (12 months induction treatment) were compared in unselected patients with UA, although there was no overall difference in progression to RA at 30 months.82 There was also no difference in disease remission rate at 30 months. However, when considering only the patients with anti-CCP-positive UA, 93% of the placebo arm progressed to RA compared with only 67% of the MTX arm (p<0.001) indicating a preventive effect. Furthermore, in a subsequent analysis restricted to 22 patients with ‘high-risk’ UA, a clear benefit of MTX was demonstrated; 6/11 (55%) of patients with high-risk UA developed RA in the MTX arm compared with 11/11 (100%) in the placebo arm, although in small numbers of patients.81 Of the patients that developed RA in the MTX arm, this was delayed compared with placebo (median 22.5 months vs 3 months; p<0.001) and DFR was achieved by 4/11 (36%) in the MTX arm compared with 0/11 (0%) in the placebo arm (p=0.031). A separate study also showed a beneficial effect of MTX in patients with anti-CCP-positive UA; after 12 months’ therapy, only 17.2% of patients in the MTX arm progressed to RA compared with 78.9% of those in the placebo arm (p<0.001).84 Boolean remission was achieved in 46.4% of the MTX arm compared with 17.6% of the placebo group (p=0.057).
Six months of ABT therapy in patients with anti-CCP-positive UA also showed beneficial effects on radiographic and MRI disease progression and anti-CCP levels at 1 year compared with placebo.121 Fewer patients in the ABT arm progressed to RA, but statistical significance was not reached in this small study. An important caveat, as with many of the UA studies, is that many of the included patients would now meet the updated classification criteria for RA58 and would no longer be considered eligible for such studies.
Trials investigating the benefits of induction therapy with corticosteroids in altering disease progression in UA have produced less impressive results.79 124 One such trial investigated the effect of three 80 mg intramuscular injections of methylprednisolone compared with placebo on disease progression (judged as the need to start DMARDs) and development of RA at 1 year. Corticosteroids delayed disease progression (76% vs 61% referred to start DMARDs at 6 months; OR 2.11, 95% CI 1.16 to 3.75, p=0.015) but did not prevent the development of RA.124 Similarly, in the SAVE trial, a single 120 mg intramuscular injection of methylprednisolone produced similar disease remission rates compared with placebo at 1 year (16.2% vs 17.8% in methylprednisolone vs placebo) and similar progression to RA (45.1% in steroid arm vs 50.1% placebo arm).79
Studies investigating the use of tumour necrosis factor (TNF) - alpha inhibitors as induction therapy in UA have been limited; those reported have failed to show an impact in altering progression to RA.68 83 In patients with high-risk UA, only 20% of IFX-treated patients achieved remission at 6 months, compared with 14% of placebo. All patients in the IFX arm developed RA at 1 year.68 The majority of patients with IFX-treated UA also progressed to RA in a recent study (73% of IFX arm compared with 67% of placebo arm).83
The limited available studies in patients with PR suggest drug intervention may have a role in altering progression to RA in this population, although the evidence is much weaker than for UA, as appropriately designed clinical trials have not been performed. Antimalarial therapy appeared to delay the development of RA (162 months vs 56 months, p=0.03) in a retrospective cohort study, although there was no difference in overall rates of progression to RA.61 Relatively low progression rates to RA were also reported in cohorts of patients with PR treated with DMARDs, although these were not controlled studies, which limits their interpretation.65 85
Considering the available data, the task force felt drug intervention should be considered in patients with UA with the aim of reducing disease progression and improving RA outcomes. The benefit of drug intervention in PR is less clear at present although antimalarials may be beneficial and warrant further study. There is no current evidence for the use of drug intervention in delaying disease progression in at-risk individuals without clinical arthritis.
9. In clinical trials and observational studies, individuals should be informed about their risk of developing RA using an approach tailored to the individual participants (LoA 9.5)
The task force felt it important that individuals at risk of RA are optimally engaged with strategies to identify their risk and/or potentially reduce their risk through intervention. Thematic synthesis of qualitative data and quantitative data from preventive intervention studies informed points to consider 9 and 10.
Only a few studies have explored the perspectives of individuals at risk of RA regarding risk prediction and RA prevention. There are two studies in people with MSK symptoms but without clinical arthritis (including arthralgia)125 126 and five in FDRs.127–132 Additionally, there is one study in people who have been diagnosed with RA133 and another includes views from members of the public.134
Individuals with arthralgia (94%) report that they have benefited from being informed about their risk of developing RA.126 FDRs are aware of their susceptibility to RA but at the same time unsure of the extent of their risk.128 FDRs have raised concern that knowing their absolute risk would increase their anxiety and potentially affect decisions about their future and they would need additional support to understand the risk and cope with the emotional impact of this information.128 A randomised controlled trial (RCT) comparing a web-based tool (Personalized Risk Estimator for Rheumatoid Arthritis) with standard, non-personalised RA education showed that the tool may help such individuals to better calculate disease risk.131
Identifying personal risk factors is important for FDRs, particularly when it comes to addressing modifiable ones such as diet.129 An RCT conducted in FDRs without RA concluded that personalised medicine approaches increase motivation for those at risk to improve behaviours that reduce the risk of developing RA.130
An individual tailored approach for communication has been acknowledged by patients with RA; they have highlighted that sharing risk information with relatives may cause negative emotions, particularly because of the negative impact on quality of life that RA has133; as such, they would prefer to choose with whom the information is communicated.
The task force agreed that it was important for individuals participating in clinical trials and observational studies to understand their personal risk of developing RA. Communication should be tailored to the individual and additional support should be considered. This is particularly important for promoting participation and engagement in prevention studies.
10. An individual’s knowledge about their risk of progression to RA may inform their decision to participate, or not, in clinical trials and observational studies (LoA 9.85)
The variable accuracy of predictive models in identifying an individual’s risk of developing RA has been raised by FDRs128 and people with CSA, highlighting difficulties in interpreting prognostic information given to them.125 People with CSA preferred to have information on the origin of their symptoms,125 thus exploring illness perceptions to guide treatment decisions, rather than risk percentages. How individuals receive information about their risk may, therefore, contribute to their decision to participate in interventional studies or not.
A recent study included at-risk individuals, defined on the basis of ACPA/RF positivity. In that study, those with arthralgia (≥one peripheral joint) were more likely to have had an autoantibody test to help identify the cause of their symptoms; in contrast, asymptomatic individuals were more likely to have had a test to contribute to research. Many symptomatic individuals expressed willingness to undergo additional predictive testing, including an assessment of the synovium by biopsy, if that would help further refine risk estimation. Asymptomatic individuals were less likely to consider further predictive testing.
Importantly, both groups highlighted the need for tailored, patient-understandable information to be delivered by an HP.
Prevention intervention studies are typically grouped into those that involve lifestyle and/or behaviour modification or those that involve taking medication. At-risk individuals with symptoms are more likely to consider both interventions.126 This contrasts with FDRs who would prefer to wait until symptoms developed before considering drug interventions.129 An understanding of personal risk is more likely to improve RA-risk-related behaviours such as dental hygiene and dietary change.129 130 However, preventive treatment offering the largest risk reduction is not necessarily the priority for asymptomatic FDRs.132
The task force acknowledged that level of baseline risk typically informs study protocols for preventive interventions, either lifestyle/behaviour modification or pharmacotherapy. Hence, supporting individuals at risk of RA to understand their personal risk factors and overall level of risk is likely to help inform their decision to participate, or not, in clinical trials and observational studies.
The task force acknowledged that points 9 and 10 follow the same theme, but convey separate messages, which justifies having two separate points. Point 9 refers to the approach to be used to inform participants about their risk. This point is centred on communication strategies, which should be considered when planning clinical trials and studies in at-risk populations. Point 10 refers to factors which may influence participation in trials and clinical studies. This point explores reasons why participation in studies may be limited and how it could be improved.
Conclusion
The goal of this EULAR task force was to provide the first expert consensus and guidance on the conduct of clinical trials and observational studies in individuals at risk of RA. These studies represent a new and evolving area in rheumatology research and clinical practice. Although much of the guidance is based on robust data from multiple studies, some is based on low levels of evidence and expert opinion. Therefore, guidance statements have been formulated as ‘points to consider’ rather than recommendations (table 1). Validation of the points to consider was beyond the scope of the task force but could be considered in a larger independent group of stakeholders in future. We acknowledge that guidance could not be provided in some specific areas, for example, at-risk individuals who develop non-articular diseases (eg, eye disease and lung disease) in the absence of arthritis. This is due to a lack of published evidence in these areas.
Several clinical trials in individuals at risk of RA are in progress and there is a growing interest from multiple stakeholders, including rheumatologists, academics, policy-makers, the pharmaceutical industry and, most importantly, patients. In many ways, these studies represent uncharted territory in rheumatology; aiming to prevent arthritis those with risk factors, rather than the conventional paradigm of suppressing the disease once it is clinically established. As such, there are many important differences and unknowns. The goal of the task force was to address these uncertainties by providing expert consensus and data-driven guidance where available to help optimise the conduct of work in this area.
The overarching principle and 10 points to consider set out a broad framework, which covers the key areas for conducting clinical trials and studies in at-risk individuals. The areas included were those prioritised by the task force. This includes the different types of at-risk populations, and how they may be distinguished based on clinical presentation, for trials and studies. For each of these populations, guidance on appropriate study end-points and trial outcomes, and the core and emerging risk factors, which should be assessed, are provided (table 3, figure 1). Considerations for optimising participation in these studies and informing at-risk individuals about their level of risk are also included. Finally, a research agenda, agreed by the task force, has also been proposed (box 1).
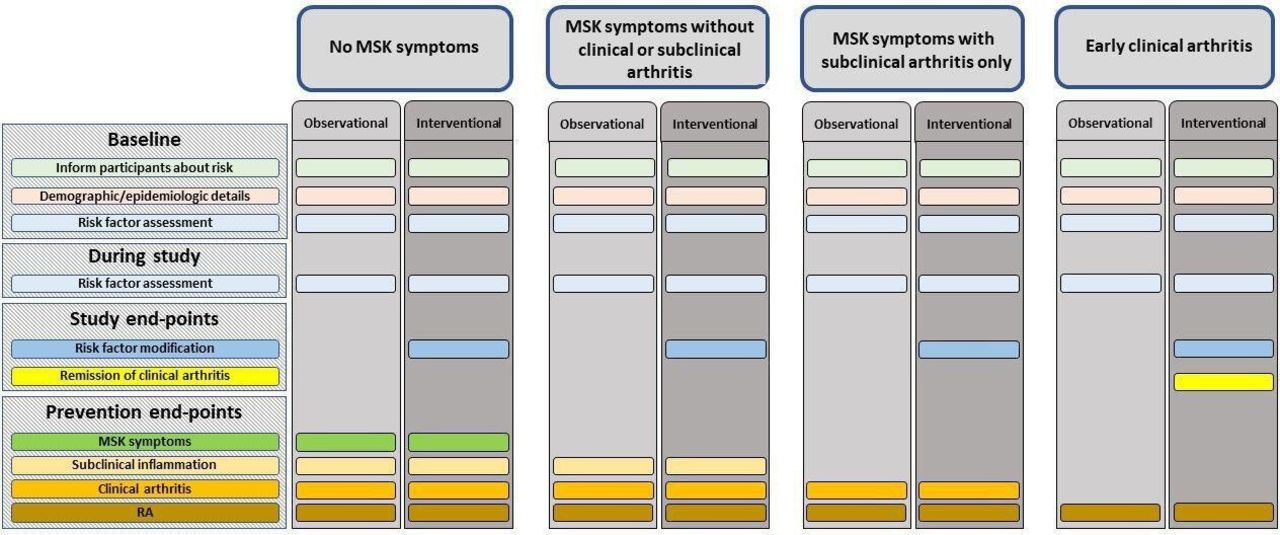
{kind=link}
A summary of assessments and end-points which should be collected in clinical trials and observational studies of individuals at risk of RA, according to the at-risk population. MSK, musculoskeletal; RA, rheumatoid arthritis.
Research agenda
The task force agreed that future research should address the following key questions:
Should individuals with mucosal inflammation/dysbiosis (periodontal, lung or gut) with or without genetic predisposition or serum autoantibodies be included as an at-risk group?
Are non-musculoskeletal symptoms (eg, stress-related symptoms and fatigue) prevalent in individuals at risk of developing rheumatoid arthritis (RA)?
Which surrogate biomarkers should be used as end-points in pilot/early phase interventional studies? And in which populations?
Improvement of subclinical inflammation on ultrasound (US)/MRI in symptomatic individuals?
Improvement of clinical features (eg, early morning stiffness duration and small joint tenderness) in symptomatic individuals?
Improvement of mucosal inflammation with or without dysbiosis in seropositive asymptomatic individuals?
Development of autoimmunity in seronegative individuals with genetic risk (including first-degree relatives)?
Improvements in patient reported outcomes (PROs) for symptomatic patients? And which patient reported outcomes should be used as end-points?
Biomarkers of inflammation/autoimmunity (emerging).
Can at-risk populations, from the background population, be cost-effectively identified, recruited and given preventative treatment based solely on demographics (age, sex and smoking status)?
Does subclinical inflammation on imaging represent a relevant endpoint (with distinct predictors)?
Do the risk factors that drive RA autoimmunity and disease progression vary according to the ethnicity or geography of the population?
Which biomarkers/risk factors change as individuals progress to inflammatory arthritis?
In individuals at risk of RA, what is the sequence and timescale of the changes in biomarkers/risk factors?
How frequently should we reassess an individual’s risk and is this subpopulation dependent?
Which biological pathways are linked with progression to RA?
Should interventions be personalised to an individual’s risk factors, for example, smoking cessation, treatment of periodontitis and weight loss?
In those at high risk, should multimodal intervention be considered according to risk factors, for example, immunomodulation combined with periodontal therapy/smoking cessation/weight loss as appropriate?
Does reduction in one or more risk factors reduce the likelihood of progression?
Can the quantification of an individual’s risk be improved, and risk scores validated?
Are interventional studies in at-risk individuals cost effective?
Should studies assess the long-term impacts of pre-arthritis interventions, including impacts on the RA phenotype (eg, severity, treatment response, DFR, etc), if it develops?
Should we develop standardised methodologies to optimise acquisition and comparability of potentially relevant clinical and epidemiological data and biospecimens (eg, blood, oral, lung, gut and synovium) in studies of individuals at risk for RA?
What is the optimum approach (including psychological support/counselling) for conveying risk of RA to these individuals?
How do at-risk individuals assess risk versus benefit in deciding on participation in either lifestyle or drug prevention studies?
Which risk factors do patients consider are high risk for developing RA?
Should risk prevention strategies be tailored for differing cultural dimensions?
These statements should help harmonise the datasets produced by future studies and facilitate collaboration in this important area. They should also improve the validity of individual trials and studies, optimising outputs from hard to recruit populations, which often require unique patient cohorts and infrastructure. It is hoped that this guidance will help galvanise future collaborative efforts in studies of at-risk individuals and RA prevention.
Ethics statements
Acknowledgments
We would like to thank Mr Joel Kerry and Dr Andrea Di Matteo for assistance with the systematic literature reviews. We would also like to thank Dr Francesco De Leonardis, Dr Jens Gammeltoft Gerwien (Eli Lilly & Co) and Ms Marie Brazil (Bristol-Myers Squibb) for taking part in the task force as industry stakeholders. Their input was based on their own personal opinions and do not represent the views of Eli Lilly & Co or Bristol-Myers Squibb.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Johannes WJ Bijlsma
Twitter @HeidiSiddle
Correction notice This article has been corrected since it published Online First. Dr Cañete's name has been corrected.
Contributors The manuscript was drafted by KM and PE who convened the task force. All other authors were members of the task force and contributed by making comments and revisions to the manuscript.
Funding Funded by EULAR grant CLI 115.
Competing interests KM: honoraria from Abbvie, Eli Lilly & Co and UCB; grants from Eli Lilly & Co and Gilead. HJS: none declared. AK: speakers bureau, consultancy: AbbVie, Bristol-Myers Squibb, Celgene, Eli Lilly & Co, Gilead, Merck Sharp and Dohme, Novartis and Pfizer. DAR: scientific advisor for GSK. AIC, JDC and APC: none declared. CID: honoraria from Roche, Chugai, Pfizer, BMS, MSD, Biogen, Abbvie, Sandoz, Janssen, Novartis, Fresenius Kabi and Sanofi; research grants from Pfizer, MSD and Schwa Medico. KR: speaker fees from Abbvie. PE: expert advice to Pfizer, Abbvie, Amgen, MSD, Roche, Sanofi, BMS, Novartis, Eli Lilly & Co, Gilead, Samsung and Celltrion; grants from Abbvie, Eli Lilly & Co, BMS and Samsung.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.