Article Text

Abstract
Objective: To evaluate the safety and efficacy of abatacept during 2 years of the ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders) trial in patients with rheumatoid arthritis.
Methods: Patients completing the 6-month, double-blind period were eligible to enter the long-term extension; patients received abatacept ∼10 mg/kg, plus disease-modifying antirheumatic drugs. Safety and efficacy (American College of Rheumatology (ACR) criteria responses, DAS28 (C-reactive protein), HAQ-DI, SF-36, Medical Outcomes Study Sleep Problems Index, fatigue VAS) were assessed through 2 years.
Results: 317 patients (218 from the abatacept and 99 from the placebo group) entered and 222 (70%) completed 18 months of long-term extension treatment. The incidence and type of adverse events were consistent between the double-blind and cumulative (double-blind plus long-term extension) periods. Rates of serious adverse events were 25.6 and 23.4 per 100 patient-years in the double-blind versus cumulative period. At 6 months and 2 years, using non-responder analyses, ACR responses in abatacept-treated patients were: ACR 20, 59.4% and 56.2%; ACR 50, 23.5% and 33.2%; ACR 70, 11.5% and 16.1%; HAQ-DI responses were 54.4% and 47.9%. At 6 months and 2 years, using post-hoc as-observed analyses, the percentage of patients (95% confidence interval) achieving DAS28 (C-reactive protein) low disease activity score (⩽3.2) and DAS28 (C-reactive protein)-defined remission (<2.6) increased from 18.3% (13.0, 23.5) to 32.0% (24.6, 39.4) and 11.1% (6.8, 15.3) to 20.3% (13.9, 26.6). Clinically meaningful improvements in SF-36, pain, fatigue and sleep problems were also maintained throughout the 2 years of abatacept treatment.
Conclusion: No unique safety observations were reported during open-label exposure. Improvements in the signs and symptoms of rheumatoid arthritis, physical function and health-related quality of life observed after 6 months, were maintained throughout the 2 years in this population with difficult-to-treat disease.
Trial registration number: NCT00124982.
Statistics from Altmetric.com
Rheumatoid arthritis (RA) therapies should provide improvements in clinical signs and symptoms as well as meaningful changes to a patient’s physical function and quality of life (QoL) that are durable over time. Standard treatments for RA include traditional disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX), and biological agents, such as tumour necrosis factor (TNF) inhibitors. While these therapeutic options have offered clinical benefits for many patients, a substantial proportion of patients do not respond to anti-TNF therapy1–4 and some may lose their response over time.5 A number of patients may also develop drug-related toxicities,1 2 4 or form antidrug antibodies6 7 to anti-TNF therapies.
Therapies have recently been introduced to treat the growing population of patients who had an inadequate response to anti-TNF therapy.8 9 These therapies target the pathogenesis of RA, via different mechanisms of action than traditional DMARDs and anti-TNF agents. Abatacept is a fully human soluble co-stimulation modulator that selectively targets the CD80/CD86:CD28 co-stimulatory signal required for full T-cell activation. In the ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders) trial, the efficacy and safety of abatacept was assessed in patients with active RA and an inadequate response to anti-TNF therapy.9 The 6-month, double-blind phase of this study provided the first evidence to support a treatment strategy in patients who have failed to respond to TNF inhibition, demonstrating significant improvements with abatacept treatment in signs and symptoms, physical function and health-related QoL (HRQoL).9
We present the safety and efficacy findings over 2 years of treatment with abatacept in patients with an inadequate response to anti-TNF therapy, including a 6-month double-blind period and an 18-month, open-label, long-term extension (LTE) in the ATTAIN trial.
PATIENTS AND METHODS
Patients
Eligible patients are those that met the inclusion criteria for the 6-month, randomised, double-blind, placebo controlled, multicentre phase of the ATTAIN trial.9 Patients had active RA and an inadequate response to anti-TNF therapy due to lack of efficacy (either etanercept or infliximab or both (administered sequentially)), as described previously,9 and had completed 6 months of treatment in order to enter the LTE. Two groups of patients were enrolled: (1) those with a previous inadequate response to ⩾3 months of anti-TNF therapy; (2) those with a current inadequate response (i.e. at enrolment) to ⩾3 months of anti-TNF therapy. Washout of anti-TNF therapies was performed prior to randomisation.9
Study design
The objectives of the LTE period were to assess the safety and efficacy of abatacept in combination with background DMARDs over 2 years of treatment. For patients originally randomised to placebo during the double-blind period, the effect of switching from placebo to abatacept was also assessed. Abatacept was administered as a 30-minute intravenous infusion and dosed according to weight as follows: patients weighing less than 60 kg, 60–100 kg or more than 100 kg received 500 mg, 750 mg or 1000 mg of abatacept, respectively. In the double-blind period, abatacept was administered on days 1, 15 and 29, and every 4 weeks thereafter. In order to maintain blinding to treatment group assignment, all patients who entered the LTE received abatacept every 4 weeks, with no additional loading dose at week 2 for patients who were switched from placebo to abatacept treatment at the end of the double-blind period. No premedication was required.
During the LTE period, patients also were required to continue to receive one or more background DMARDs or anakinra, adjustments of which were permitted at the discretion of the investigator based on the patient’s clinical status. Patients were not allowed to resume anti-TNF therapy. Adjustments in other concomitant medications also were permitted at the discretion of the investigator.
Both the double-blind and the LTE periods were approved by the appropriate Institutional Review Boards/Independent Ethics Committees at participating sites, and were conducted in accordance with the ethical principles of the Declaration of Helsinki. All patients provided written informed consent. The Food and Drug Administration registration number for this clinical trial is NCT00124982.
Safety assessments
Safety assessments were performed monthly and included: all reported adverse events (AEs) and serious AEs (SAEs; including deaths); discontinuations due to AEs, clinically significant changes in vital signs, physical examinations and clinical laboratory test abnormalities; and immunogenicity evaluations. AEs and SAEs were classified by system organ class and preferred term using the Medical Dictionary for Regulatory Activities (MedDRA) classification, version 8.0 (previously published safety data from the ATTAIN trial over 6 months were generated using the MedDRA version 7.0 classification system).
Clinical assessments
Evaluations for changes in signs and symptoms of RA, physical function and HRQoL were performed quarterly during the LTE period on days 253, 365, 449, 533, 617 and 729. For both treatment arms (patients originally randomised to abatacept, and patients originally randomised to placebo who then switched to abatacept at the end of the double-blind period) baseline values were recorded on day 1 of the double-blind period.
American College of Rheumatology responses
Clinical efficacy was measured using the American College of Rheumatology (ACR) criteria.10 The percentage of patients achieving an ACR 20, 50 and 70 response10 was measured at each visit. The proportions of patients achieving a major clinical response, defined as the maintenance of an ACR 70 response over a continuous 6-month period, and an ACR 70 maintained for a continuous 9-month period in the same patient, were also evaluated.
Disease activity score 28 assessments
Improvements in disease activity were evaluated using the Disease Activity Score 28 (DAS28; based on C-reactive protein (CRP) levels). The percentage of patients achieving a low disease activity state (LDAS; DAS28 (CRP) ⩽3.2) and DAS28 (CRP)-defined remission (DAS28 (CRP) <2.6)11 were determined.
Physical function and health-related quality of life
Improvements in physical function were measured using the Health Assessment Questionnaire-Disability Index (HAQ-DI).12 The percentage of patients achieving an improvement of ⩾0.3 in the HAQ-DI (a clinically meaningful improvement, exceeding the minimum clinically important difference of ⩾0.22) was assessed.12 Health-related QoL was assessed using the Short Form (SF)-36, and an improvement from baseline of ⩾3 units was considered clinically meaningful.13 Improvements in pain, sleep quality and fatigue also were evaluated. Pain was assessed on a 100 mm visual analogue scale (VAS).14 Sleep quality was assessed using the Medical Outcomes Study Sleep Problems Index (MOS-SPI).15 The patients’ assessment of fatigue severity was measured using a fatigue 100 mm VAS.16
Statistical analysis
Baseline demographics, clinical characteristics and RA clinical history were analysed descriptively for all patients who entered the LTE period.
All patients who received at least one infusion of study drug were assessed for safety.
Data presented for the cumulative study period (double-blind plus open-label periods) are based on all patients originally randomised to abatacept and who received at least one dose of abatacept, plus all patients originally randomised to placebo and who entered the LTE (and subsequently received one dose of abatacept). Incidence rates and frequencies were calculated for AEs, SAEs, infections and serious infections. Rarer events (such as malignancies and autoimmune events) were described using frequencies only. Incidence rates were presented per 100 patient-years of exposure, and were calculated as the number of patients with the event of interest (numerator) divided by the total exposure (denominator) for the specified treatment period. A patient’s contribution to exposure ended at the time of the first occurrence of the event of interest.
Patients who received at least one infusion of abatacept during the LTE period (the intent-to-treat (ITT) population) were included in the efficacy analyses. Efficacy end points were assessed by either protocol pre-specified or post-hoc as-observed analyses. The protocol pre-specified analyses were performed on the ITT population using either data from all patients, with those who discontinued considered as non-responders (non-responder analysis), or using only patients with data available at the visit of interest (as-observed analysis). Additional post-hoc analyses were performed on the ITT population using as-observed data.
Non-responder analyses of ACR and HAQ-DI were performed, as well as post-hoc as-observed analyses. Protocol pre-specified analyses of the following measures were performed using as-observed data: DAS28 (CRP) mean change, SF-36, pain, fatigue and sleep quality. Post-hoc as-observed analyses of LDAS and DAS28 (CRP)-defined remission were also performed. Assessments of a major clinical response and the proportion of patients maintaining an ACR 70 for 9 continuous months were assessed using non-responder analyses. Efficacy assessments were summarised by original randomisation group; however, no formal statistical comparisons were performed between the treatment groups.
RESULTS
Patient baseline demographics and clinical characteristics
At randomisation, demographic and baseline clinical characteristics for all patients entering the LTE period were comparable across treatment groups (table 1); most patients were female, with a mean disease duration of approximately 11–12 years.
Patient disposition
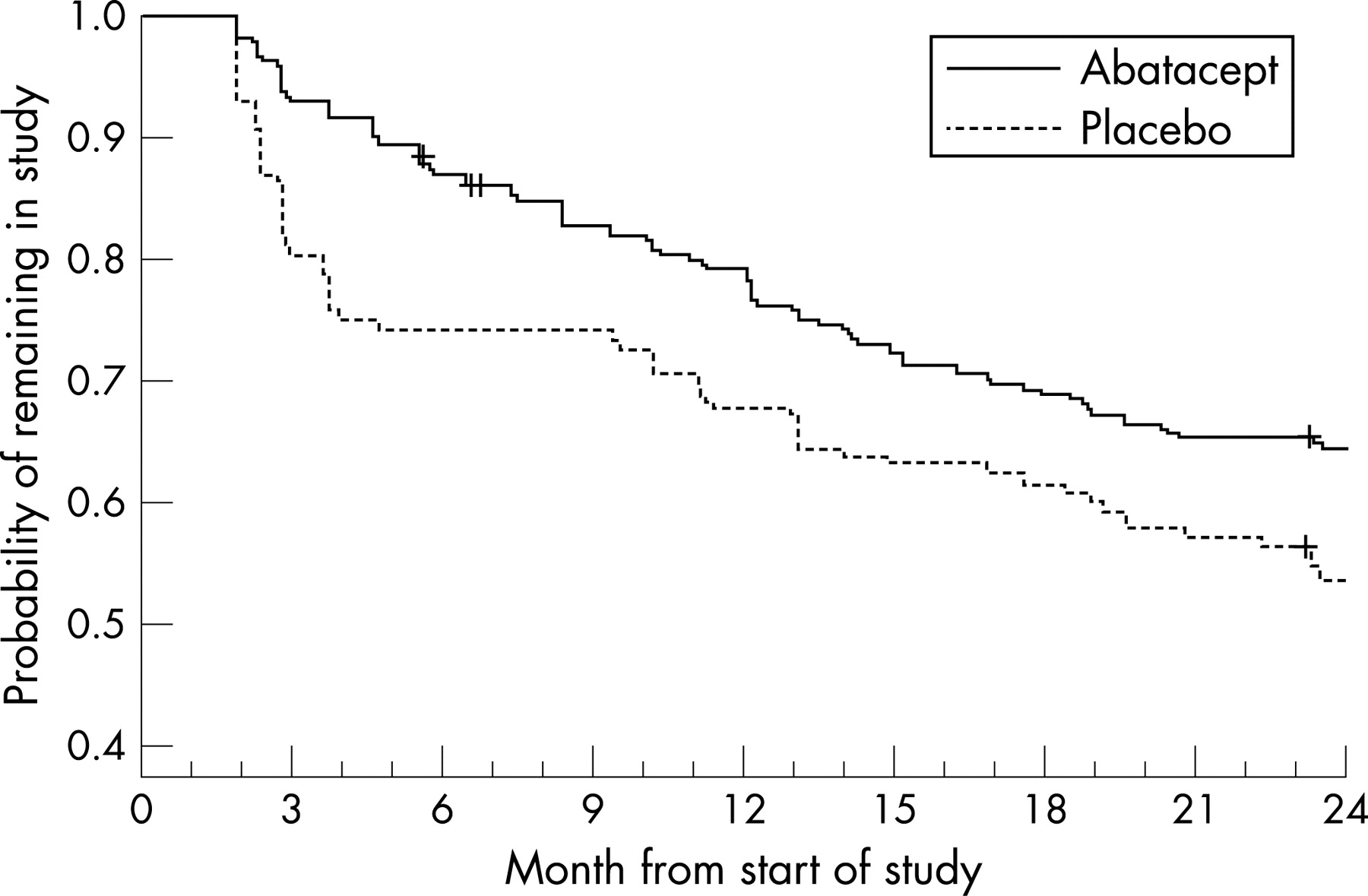
Of the 258 and 133 patients randomised and treated with abatacept and placebo during the double-blind period, a total of 218 (84.5%) and 99 (74.4%) patients, respectively, completed 6 months and entered the LTE period (fig 1). A total of 222 (of 317 who entered the LTE) patients (70.0%) were still participating in the study following 18 months of the LTE. The most common reasons for discontinuation were lack of efficacy (52 patients; 16.4%) and AEs (24 patients; 7.6%). The 52 patients who discontinued due to lack of efficacy included 34 patients from the original abatacept group (34 of 218, 15.6%) and 18 patients from the original placebo group (18 of 99, 18.2%) (fig 1). A Kaplan–Meier plot showing the probability of patients remaining in the trial throughout the 2 years is presented in fig 2.

For the original abatacept and placebo groups, respectively, MTX (79.4% and 84.8%), non-steroidal anti-inflammatory drugs (80.3% and 80.8%) and corticosteroids (82.1% and 73.7%) were the most frequently used antirheumatic medications taken concomitantly with abatacept at any time during the LTE period. For patients originally randomised to abatacept, the mean number of concomitant DMARDs, the proportion of patients receiving prednisone, and the mean dose of MTX and prednisone remained similar throughout 2 years. The mean number of DMARDs was 1.2 at baseline, 6 months and 2 years. The percentage of patients receiving prednisone was 59.2%, 59.6% and 49.7% at baseline, 6 months and 2 years, respectively. The mean MTX dose was 14.9, 15.2 and 15.1 mg/week, and the mean prednisone dose was 7.8, 7.5 and 7.3 mg/day at baseline, 6 months and 2 years, respectively.
Safety
Overall safety summary
During the double-blind period, a similar frequency of both AEs and SAEs was observed in the abatacept and placebo groups throughout 6 months of treatment.9 A summary of the safety results for patients who received at least one dose of abatacept, for the double-blind versus the cumulative (double-blind plus LTE) study periods is presented in table 2. The types of AEs, including infusional events, were generally similar to those reported during the double-blind period.9
Incidence rates of SAEs were consistent for the double-blind and cumulative periods, with rates of 25.6 and 23.4/100 patient-years, respectively. Overall, a total of 172 SAEs were reported in 103 patients during the cumulative study period. The most frequently occurring events (excluding those related to arthritis) classified by category were infections and infestations (25 patients; 5.0/100 patient-years) and neoplasms, including those classified as benign, malignant and unspecified (12 patients; 2.3/100 patient-years).
During the cumulative period, discontinuations due to AEs and SAEs occurred in 25 and 18 patients, respectively. There was no pattern in the AEs that led to discontinuation; no AE leading to discontinuation was reported by more than two patients. A total of two deaths occurred during 2 years of the ATTAIN trial; both being in abatacept-treated patients. One patient died of myocardial infarction and congestive heart failure on day 104 of the double-blind period, and one case of Enterobacter pneumonia resulted in death on day 569 of the LTE.
Infections
Throughout the cumulative period, overall infections and serious infections occurred in 234 and 25 patients, respectively, with incidence rates of 89.4 and 5.0/100 patient-years, respectively. These rates were similar to those reported during the double-blind period, during which incidence rates of 108.8 and 5.3/100 patient-years were reported, respectively. The most frequent serious infections (occurring in >0.5% patients) during the cumulative period were recorded as pneumonia and lobar pneumonia. Discontinuations due to infections during the cumulative period occurred in five patients; three of these infections were serious (pneumonia, streptococcal sepsis and subacute endocarditis).
Malignancy
A total of 11 malignant neoplasms were reported in the cumulative period. The most frequently reported malignant events were non-melanomatous skin cancers; basal cell carcinoma in three patients and squamous cell carcinoma in two patients. The remaining events were each reported by one patient: breast cancer, ovarian epithelial cancer, prostate cancer, T-cell lymphoma, metastatic thyroid cancer, thyroid neoplasm and uterine cancer.
Autoimmune symptoms and disorders
Autoimmune symptoms and disorders were reported in 15 patients in the cumulative period. This included four cases reported in the double-blind period and 11 cases reported during the 18 months of open-label treatment. The only events reported by more than one patient were psoriasis (in three patients) and vasculitis (in two patients).
Clinical efficacy
Improvements in the signs and symptoms of rheumatoid arthritis
Improvements in signs and symptoms observed at 6 months were maintained through 2 years in patients originally randomised to abatacept (at 6 months and 2 years using the non-responder analyses: ACR 20, 59.4% and 56.2%; ACR 50, 23.5% and 33.2%; ACR 70, 11.5% and 16.1%, respectively) (fig 3A). When switched to receive abatacept, patients in the original placebo group achieved comparable improvements at 2 years with patients in the original abatacept group (using the non-responder analysis, ACR 20, 50 and 70 responses for patients in the original placebo group were 51.5%, 32.3% and 13.1%, respectively; fig 3B).
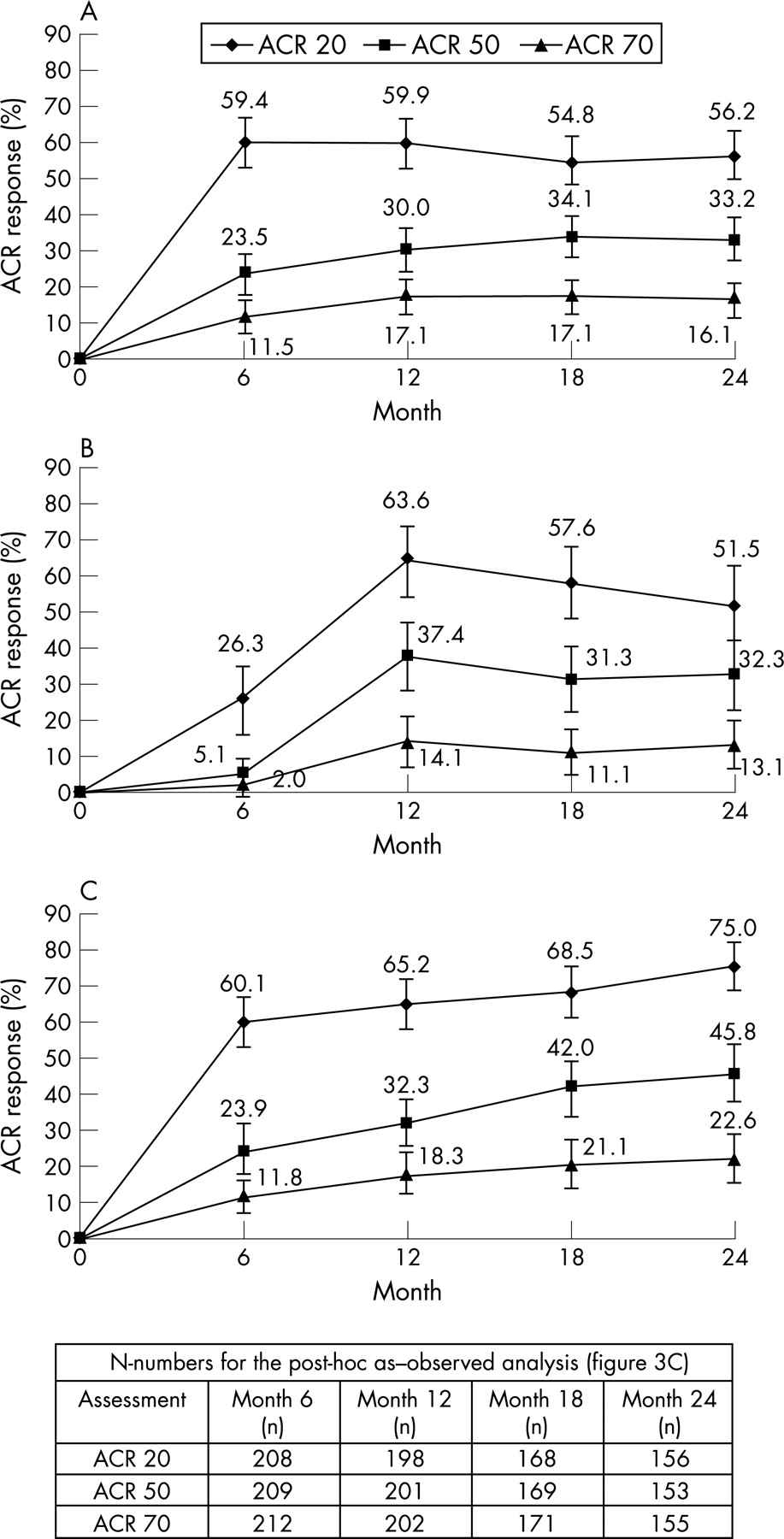
Figure 3C presents the post-hoc as-observed ACR response data over 2 years for patients originally randomised to abatacept. As with the non-responder analyses, improvements observed at 6 months were maintained throughout the 2 years. For ACR 20 and 50, 95% CIs do not overlap at 6 months and 2 years.
For patients in the original abatacept and placebo groups at 2 years, the percentage of patients achieving a major clinical response was 18.9% and 16.2%, respectively, with 10.6% and 5.1% demonstrating a maintained ACR 70 for 9 continuous months.
Disease activity
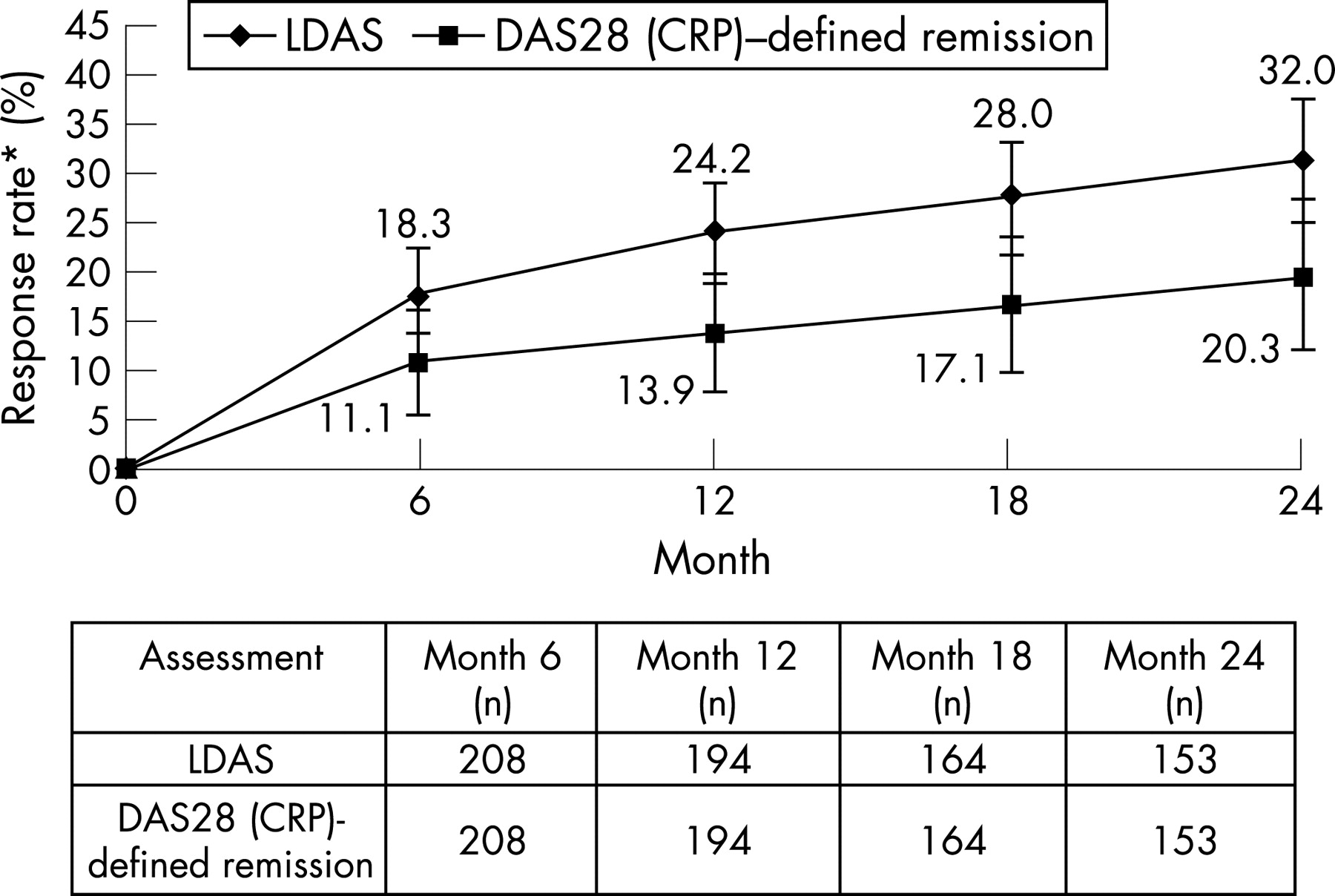
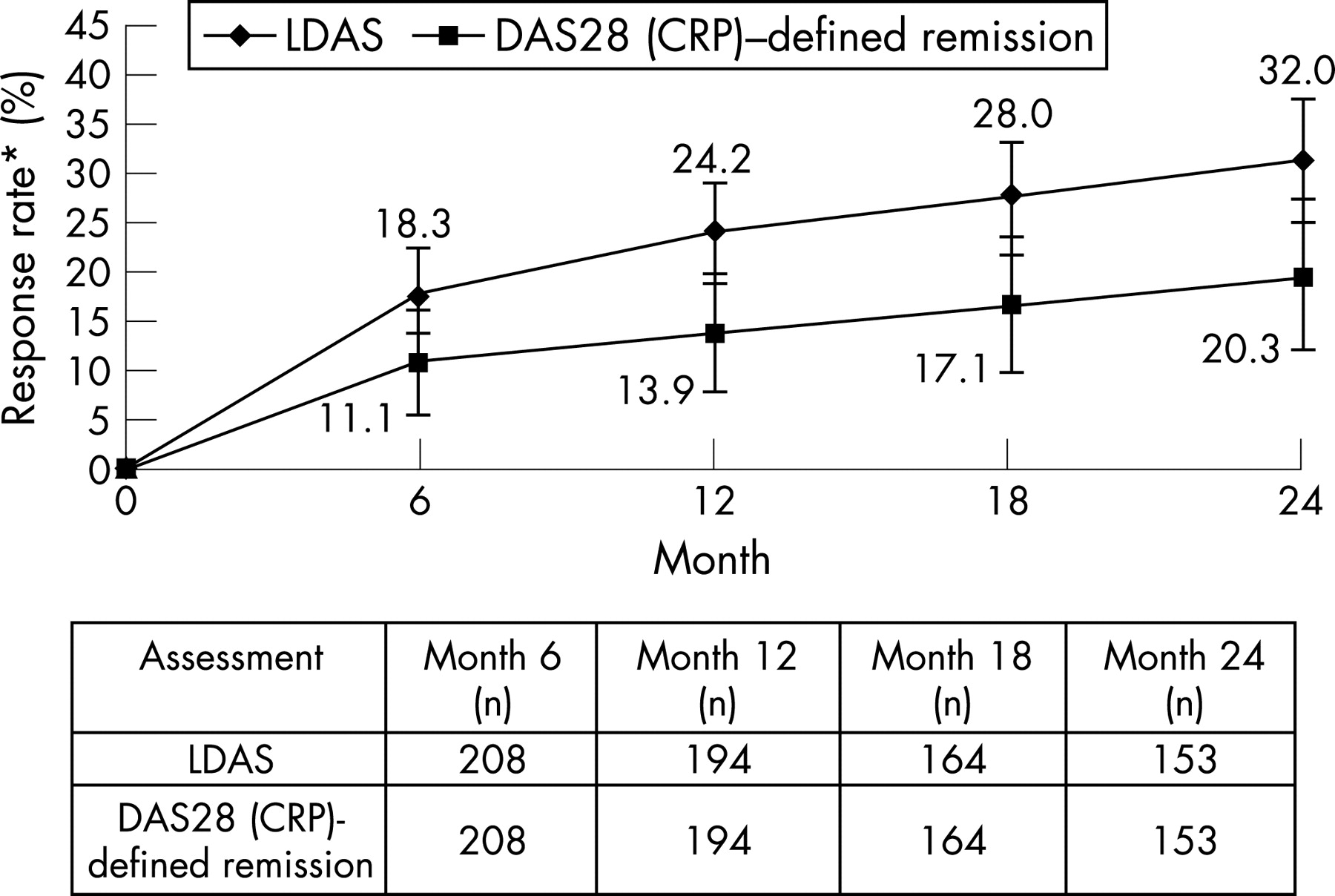
At 2 years, the mean change from baseline in DAS28 (CRP) for patients originally randomised to abatacept was −2.66 (standard error: 0.11), providing further reductions in disease activity than observed at the end of the double-blind period (−1.99 for the original abatacept group). Using the post-hoc as-observed analyses, the percentage of patients originally randomised to abatacept (95% CI) who achieved LDAS and DAS28 (CRP)-defined remission was shown to increase from 18.3% (13.0, 23.5) and 11.1% (6.8, 15.3) at the end of the double-blind phase to 32.0% (24.6, 39.4) and 20.3% (13.9, 26.6), respectively, at 2 years (fig 4). No overlap existed in the 95% CIs for LDAS between 6 months and 2 years.
Improvement in physical function
For the non-responder analyses, clinically meaningful HAQ-DI responses were observed for 47.9% of patients originally randomised to abatacept at 2 years, maintaining the improvements of 54.4% observed at 6 months. For patients in the original placebo group, comparable responses were observed at 2 years (47.9% vs 46.5%, for the original abatacept versus placebo groups, respectively). For the post-hoc as-observed analyses, a similar trend of maintained response was observed over 2 years of abatacept treatment (fig 5A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Improvement in health-related quality of life
The clinically meaningful improvements observed with abatacept at 6 months in the SF-36 (including the physical (PCS) and mental component summary (MCS), and all eight subscales9), were maintained over 2 years in patients originally randomised to abatacept (fig 5B,C). The greatest improvements were observed in the physical component subscales, with marked benefits in the bodily pain and role-physical dimensions. Mean improvements from baseline in the PCS and MCS summary scores at 6 months and 2 years are presented for patients in the original abatacept and placebo groups in fig 5C. For patients randomised to abatacept, the mean change from baseline in the PCS and MCS and the individual subscales at 6 months and 2 years exceeded the clinically meaningful improvement of 3 units.13 Following 2 years of abatacept treatment, mean SF-36 scores for the PCS and MCS were 38.1 and 48.3, respectively, denoting 10.3 and 6.2 points of improvement from baseline.
At 2 years, mean reductions from baseline (standard errors) in pain, sleep problems and fatigue VAS were −37.2 (2.4), −12.7 (1.4) and −28.2 (2.1), respectively, for patients originally randomised to abatacept, compared with −30.8 (2.0), −11.1 (1.1) and −25.0 (2.0), respectively, at 6 months. Improvements exceeded the minimal clinically important difference of 10 for fatigue and pain, and 6 for sleep problems.17 For patients originally randomised to placebo, reductions in pain, sleep problems and fatigue comparable with the original abatacept group were observed at 2 years; −32.5 (3.6), −12.0 (2.1) and −21.5 (3.7), respectively.
DISCUSSION
Despite the efficacy of TNF inhibitors, a substantial proportion (up to 30%) of patients with RA are refractory to anti-TNF therapy1–3 18 19 and, until recently, no proven therapies were available for this group of patients. The ATTAIN trial was the first clinical trial to assess the efficacy of a biological therapy in anti-TNF inadequate responders who discontinued anti-TNF therapies due to lack of efficacy. This LTE study period provides long-term data demonstrating consistent safety and maintained clinical benefit in this difficult-to-treat patient population.
The safety profile of abatacept through the 2-year cumulative ATTAIN trial period was comparable with that observed in the 6-month double-blind period. The overall pattern and intensity of AEs during the cumulative period was not different from that observed during the double-blind period,9 with no unique safety observations reported through open-label exposure to date. While 11 malignancies were reported during this 2-year study, the most frequent events were non-melanomatous skin cancers. The remaining malignancies were singular events, including one T-cell lymphoma. It is unclear if the malignancies were related in any way to the use of abatacept, previous therapies, or the long-standing and refractory nature of these patients’ disease; follow-up of this patient population over a longer time period will allow further assessment.
Efficacy data from this study were assessed by either non-responder analyses or post-hoc as-observed analyses. The non-responder analysis (where all patients who discontinued were considered non-responders) is considered the most conservative approach; however, it becomes less meaningful with increasing exposure. As-observed analyses may be more relevant over the long term, following only those patients who actually continue therapy in the study.
During 2 years of treatment in the ATTAIN trial, ACR responses and HAQ-DI responses were maintained through open-label abatacept treatment, regardless of the type of analysis used. A possible increasing proportion of abatacept-treated patients achieved LDAS over 2 years, while the proportion achieving DAS28 (CRP)-defined remission was maintained with the as-observed analysis. At 2 years, one-fifth of patients in the original abatacept group had achieved DAS28 (CRP)-defined remission.
Beyond providing clinical benefits for patients, RA therapies should aim to improve HRQoL. In this trial, approximately two-thirds of abatacept-treated patients experienced a clinically meaningful improvement in physical function at 2 years. The improvements observed in HRQoL also were clinically meaningful for both summary scores and all eight individual subscales and were maintained or increased from 6 months to 2 years. Combined with the maintained improvements observed in pain, sleep and fatigue, these data support the ability of abatacept to provide real and meaningful benefits to patients. Over the 18-month LTE period of this trial, patients who were originally randomised to placebo and were reallocated to abatacept experienced efficacy comparable with patients in the abatacept group. At 2 years, patients in this group experienced similar benefits in both efficacy and HRQoL outcomes to those patients treated with abatacept for the study duration.
The findings reported here should be interpreted within the context of the study limitations.
As these data are currently limited to 2 years of treatment, further follow-up of this population is required to determine longer-term safety, and to examine how long the durability of response can be maintained.
A total of 16.4% of patients discontinued during the LTE period due to lack of efficacy. Although the attrition rate appeared to increase throughout the LTE period, it should be noted that 70% of this patient population who had previously failed to respond to at least one or more anti-TNF therapies remained on treatment at 2 years and are ongoing in the LTE.
By providing maintained improvements in clinical outcomes and HRQoL combined with a consistent safety profile, abatacept provides meaningful and durable benefits to this unique and previously difficult to treat population of patients. The findings of this trial, therefore, support the benefit of selective co-stimulation modulation with abatacept in patients with active RA and an inadequate response to anti-TNF therapy.
Acknowledgments
This study was funded and sponsored by Bristol-Myers Squibb, Princeton, NJ, United States. Support for the study at Stanford University provided in part from a grant (5 M01 RR000070) from the National Center for Research Resources, National Institutes of Health. We would like to thank Laura Gardiner, MSc, Medicus International, for her editorial assistance. Editorial support was funded by Bristol-Myers Squibb, Princeton, NJ, USA. M. C. Genovese has received grant support from Bristol-Myers Squibb to conduct this and other clinical studies and has served as a consultant for Bristol-Myers Squibb. M. Schiff has received research grants and consulting fees from Bristol-Myers Squibb and Centocor. M. Luggen has received financial support from Bristol-Myers Squibb for work done on this and other clinical trials with abatacept. J-C. Becker is an employee of Bristol-Myers Squibb, with stock options and restricted shares. R. Aranda is an employee of Bristol-Myers Squibb and owns stocks. J. Teng is an employee of Bristol-Myers Squibb. T. Li is an employee of Bristol-Myers Squibb and own stocks and shares. N. Schmidley is an employee of Bristol-Myers Squibb and owns shares. M. LeBars is an employee of Bristol-Myers Squibb and holds stocks in the company. M. Dougados has received research grants, consulting fees and been on the speakers bureau for Bristol-Myers Squibb, Abbott and Wyeth, he has also received research grants and consulting fees from Centocor and Schering Plough.
REFERENCES
Footnotes
Competing interests: None.
The Food and Drug Administration registration number for this clinical trial is NCT00124982.