Article Text

Abstract
Objective To investigate the effects of BET bromodomain protein inhibition on inflammatory activation and functional properties of rheumatoid arthritis synovial fibroblasts (RASF).
Methods The expression of the BET bromodomain proteins BRD2, BRD3 and BRD4 was analysed in synovial tissue by immunohistochemistry. RASF were stimulated with tumour necrosis factor (TNF)-α, interleukin (IL)-1β and toll-like receptor (TLR) ligands (Pam3, pIC and lipopolysaccharide (LPS)) in the presence or absence of the BET inhibitor I-BET151, or siRNA targeting BRD2, BRD3 and BRD4. RASF expression of inflammatory mediators, including MMP1, MMP3, IL-6 and IL-8, was measured by q-PCR, q-PCR array and ELISA. Cellular viability, apoptosis, proliferation and chemoattractive properties of RASF were investigated using MTT, cell apoptosis ELISA, BrdU-based proliferation and transwell migration assays.
Results BRD2, BRD3 and BRD4 proteins were detected in rheumatoid arthritis (RA) synovial tissue, expressed in both RASF and macrophages. I-BET151 suppressed cytokine and TLR ligand-induced secretion of MMP1, MMP3, IL-6 and IL-8, and mRNA expression of more than 70% of genes induced by TNF-α and IL-1β. Combined silencing of BRD2, BRD3 and BRD4 significantly reduced cytokine and TLR ligand-induced expression of a subset of gene products targeted by I-BET151, including MMP1, CXCL10 and CXCL11. I-BET151 treatment of RASF reduced RASF proliferation, and the chemotactic potential for peripheral blood leucocytes of RASF conditioned medium.
Conclusions Inhibition of BET family proteins suppresses the inflammatory, matrix-degrading, proliferative and chemoattractive properties of RASF and suggests a therapeutic potential in the targeting of epigenetic reader proteins in RA.
- Rheumatoid Arthritis
- Fibroblasts
- Inflammation
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease, characterised by chronic inflammation of the joints with severe pain and swelling, joint damage and disability, which leads to joint destruction and loss of function. Despite extensive research efforts, the underlying cause for RA is still unknown and current therapies are mostly efficacious in controlling symptoms, but subsets of patients remain refractory to multiple therapies, including anti-tumour necrosis factor α (anti-TNF-α) therapy. Joint resident RA synovial fibroblasts (RASF) have emerged as key players in the pathogenesis of RA, characterised by their ability to secrete cytokines, chemokines and joint damaging enzymes and thereby actively contribute to the inflammatory state and joint damage in RA. The inhibition of apoptosis in RASF and the recruitment of inflammatory cells including macrophages and lymphocytes by RASF are major contributors to the excessive hyperplasia of the synovial tissue in RA joints. In addition, RASF have invasive and migratory properties that lead to a spreading of the disease to unaffected joints.1
We and others have identified epigenetic changes including DNA methylation, histone acetylation and other histone modifications that are associated with the intrinsic activation and the aggressive phenotype of RASF.2 ,3 For example, recent studies have identified distinct DNA methylation patterns regulating inflammatory gene expression in synovial fibroblast (SF) obtained from RA and osteoarthritis (OA) patients.4 ,5 An increasing number of studies are now providing evidence that proteins which attach (‘writers’), remove (‘erasers’) or recognise (‘readers’) specific epigenetic modifications on histones are emerging as key players in the regulation of gene expression in RA, particularly with regard to histone acetylation.6 Inhibitors of histone deacetylases, enzymes which remove acetyl modifications from histones, have been demonstrated to have anti-inflammatory properties in RASF, macrophages, peripheral blood mononuclear cells and synovial biopsy explants,7–9 although the precise mechanism(s) of action have so far not been fully addressed.
Bromodomain proteins contain one or more conserved acetyl-lysine binding domain(s) that specifically recognise(s) ε-N-lysine acetylation motifs, a key post-translational modification in the regulation of gene expression.10 ,11 Some bromodomain proteins recruit histone acetyl transferases (HATs) to specific promoter sites and contain an instrinsic enzymatic HAT domain. Therefore, bromodomain proteins play an important role in chromatin reorganisation and transcriptional activation.12 In the human proteome, 61 bromodomains have been identified in 46 diverse proteins.10 ,11 Of special interest is a subgroup of double bromodomain-containing proteins, called BET proteins. A functional role of BET proteins in different cancers and the regulation of adipogenesis has already been well established.13 However, recent studies have also provided initial evidence that BET family members also play important roles in inflammatory responses. BET bromodomain inhibitors, acting as acetyl-histone mimetics and targeting the BET proteins (BRDT, BRD2, BRD3 and BRD4), suppress the expression of pro-inflammatory cytokines and chemokines in lipopolysaccharide (LPS)-stimulated macrophages in vitro, and protect mice against sepsis in vivo.14 ,15 BET inhibitors also protect against pathology in murine models of chronic immune-mediated inflammatory disease, including experimental autoimmune encephalitis and collagen-induced arthritis.16 ,17 Based on these studies and interest in how epigenetic regulatory proteins contribute to the activated phenotype of RASF, we examined the therapeutic potential of BET protein inhibition on modification of gene expression and cellular function in RASF.
Materials and methods
Patient samples and cell preparation
SF were derived from synovial tissue specimens that were obtained from RA and OA patients during joint replacement surgery (Department of Orthopedic Surgery, Schulthess Clinic Zurich, Switzerland), and from RA patients by needle arthroscopy (see online supplementary table S1), as previously described.18 All patients fulfilled the criteria for the classification of RA and OA, respectively.19 ,20 Informed consent was obtained from all patients prior to inclusion in the study, and the study was approved by the medical ethics committees of Zurich, Switzerland, and the Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands. Cells were cultured as described elsewhere and used between passages 4 and 8 for all experiments.21
Immunohistochemistry
After deparaffinisation, tissue sections of RA and OA patients were pretreated with citrate buffer (10 mM sodium citrate, pH 6.0). Endogenous peroxidase activity was disrupted with 3% H2O2. Slides were permeabilised with 0.1% Triton in phosphate buffered saline (PBS) for 30 min. Non-specific protein binding was blocked with 5% goat serum in Antibody diluent (Dako) for 40 min. Monoclonal rabbit anti-BRD2 antibodies (1:500, abcam), polyclonal mouse anti-BRD3 (1:50, Abgent), or monoclonal rabbit anti-BRD4 antibodies (1:250, abcam) and rabbit IgG1 (isotype control) were applied overnight at 4°C. Slides were washed in PBS-T (0.05% Tween 20 in PBS) and incubated with biotinylated goat anti-rabbit (1:1000) or goat anti-mouse (1:2000; Jackson ImmunoResearch) antibodies. The signal was amplified with ABC reagent for peroxidase (Vector Laboratories) and detected with 3,3′-diaminobenzidine (Vector Laboratories). For double staining, slides were stained with either the fibroblast marker prolyl 4-hydroxylase β or CD68 the day after staining for BRDs. For staining with monoclonal mouse anti-CD68 antibodies (1:20, Dako), slides were pretreated with trypsin (1 mg/mL). Anti-CD68 antibodies and mouse IgG1 (isotype control) were applied overnight at 4°C. For staining with fibroblast marker, slides were incubated with monoclonal mouse antiprolyl 4-hydroxylase β antibodies (1:20, Acris) and mouse IgG1 (isotype control) overnight at 4°C. Slides were washed in PBS-T and incubated with biotinylated goat antimouse antibodies (1:2000). The signal was amplified with ABC reagent (Vector Laboratories) and detected with either Vector blue (Vector Laboratories) or HistoGreen (Linaris).
Statistical analysis
Statistical analysis on data sets was carried out by using the GraphPad Prism program (GraphPad Software, San Diego, California, USA). Data sets involving I-BET dose-dependency and impact of the time of administration were analysed by analysis of variance (ANOVA) with Bonferroni correction. Data of qPCR arrays were log transformed and analysed by ratio t test. Viability data were analysed by Friedman test followed by post hoc Dunn's test and comparison between effects of I-BET and JQ1 by Pearson correlation. Other differences between experimental groups were analysed by paired t test. Data are reported as means±SDs. p Values <0.05 were considered statistically significant.
Additional details of materials and methods for ELISA assays, quantitative real-time PCR assays, RASF stimulation, RASF transfection, western blotting, assessment of RASF viability, apoptosis and proliferation, and leucocyte migration assays have either been described previously and/or are described in online supplementary materials and methods.22 ,23
Results
Expression of BET bromodomain proteins
Since the BET bromodomain proteins BRD2, BRD3 and BRD4 were reported to play a functional role in controlling inflammation,14 we analysed their expression in synovial tissues derived from RA and OA patients. Nuclear staining (brown) for BRD2, BRD3 and BRD4 was similarly detected in synovial tissues derived from RA patients and OA patients (figure 1A). Double staining of BRD2, BRD3 and BRD4 with CD68 or a fibroblast marker revealed that BET proteins are present in both fibroblasts and macrophages (figure 1B). Whereas BRD3 was not detected in lymphocyte infiltrates at all, lymphocytes were positive for BRD4 and partially positive for BRD2 (figure 1C).
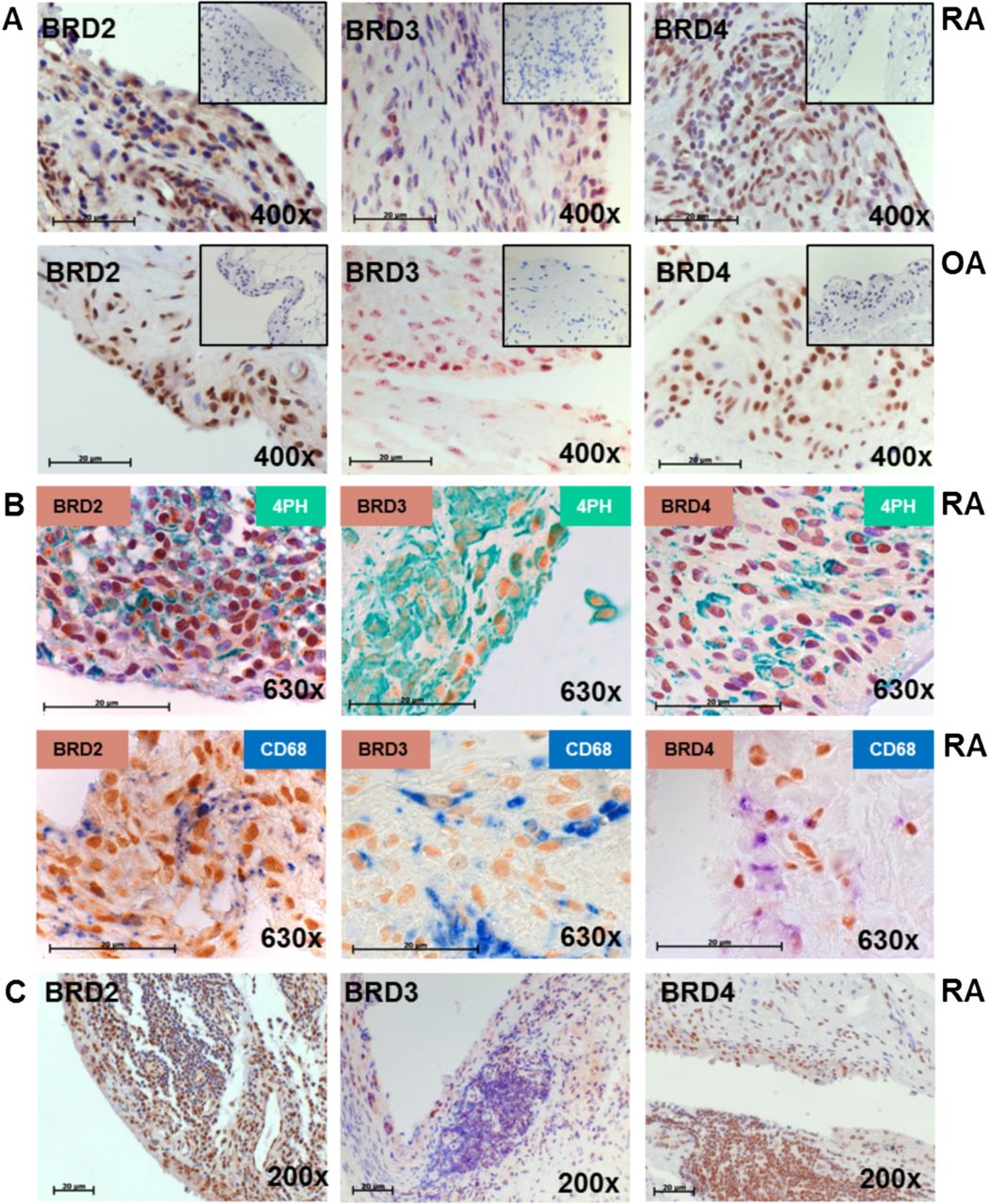
Synovial expression of BET family proteins in rheumatoid arthritis (RA). (A) Synovial tissues from RA and osteoarthritis (OA) patients were stained with anti-BRD2 (brown), anti-BRD3 (brown) and anti-BRD4 (brown) antibodies or isotype control (box). Nuclei were counter stained with hemalaun (blue). Magnification 400×. (B) Synovial tissues were double stained with anti-BRD2 (brown), anti-BRD3 (brown) and anti-BRD4 (brown) antibodies and antiprolyl 4-hydroxylase β antibodies as fibroblast marker (green) or anti-CD68 (blue) antibodies. Magnification 630×. (C) Synovial tissues with lymphocyte infiltrates present were stained as described in (A). Magnification 200×. Size bars 20 µm.
I-BET151 suppresses the expression of cytokines and MMPs by RASF in response to TNF-α and IL-1β
In order to investigate whether BET bromodomain proteins play a role in the aggressive behaviour of RASF, we stimulated RASF with TNF-α or interleukin (IL)-1β in the presence or absence of the BET bromodomain inhibitor I-BET151. I-BET151 significantly reduced MMP1 secretion after TNF-α (71.5%) and IL-1β (75.6%) stimulation, and similar and significant reductions in MMP3, IL-6 and IL-8 production were also observed (figure 2A). In the absence of RASF stimulation, I-BET151 had no effect on basal production of MMP1 or MMP3, but reduced basal IL-6 and IL-8 secretion rates by 79.4% and 39.4%, respectively (p<0.001) (figure 2A). I-BET151 effects on basal, as well as TNF-α and IL-1β-induced IL-6 (figure 2B) and IL-8 production (data not shown), were dose-dependent with 50% inhibition observed between 250 and 500 nM I-BET151. To determine if I-BET151 could impact upon inflammatory cytokine production in preactivated RASF, cells were treated with I-BET151 1 h prior to (prophylactic), 1 h after (early) or 4 h (late) after stimulation with TNF-α or IL-1β. Delaying administration of I-BET151 only slightly, but significantly, diminished the efficacy of IL-6 and IL-8 production in response to IL-1β, but not TNF-α (figure 2C).

BET family proteins regulate MMP and cytokine production in rheumatoid arthritis synovial fibroblasts (RASF). RASF (n=12) were treated with (A) TNF-α or IL-1β, or left unstimulated for 24 h in the presence or absence (control (ctrl), white bars) of I-BET151 (I-BET, 1 μM, black bars). The concentration of MMP1, MMP3, IL-6 and IL-8 in tissue culture supernatants was analysed by ELISA. (B) Dose-dependency of I-BET151 (I-BET) effects on unstimulated, TNF-α and IL-1β-stimulated RASF (n=6) IL-6 secretion as assessed by ELISA. (C) Effects of I-BET151 treatment on RASF (n=6) IL-6 and IL-8 secretion when administered 1 h before (prophylactic), 1 h after (early) or 4 h after (late) stimulation with TNF-α, and IL-1β. Data in B and C are presented as per cent of control RASF not treated with I-BET151. *p<0.05, **p<0.01, ***p<0.001.
Selective regulation of RASF gene expression by BET family inhibition
In order to assess more globally I-BET151 effects on gene transcription in RASF, we compared expression of 84 previously validated target genes regulated by TNF-α and IL-1β in RASF after cytokine stimulation in the presence or absence of the inhibitor (see online supplementary figure S1). RASF exposure to I-BET151 caused a more than 25% downregulation in the message levels of 70.2% and 73.1% of genes induced (more than twofold) by TNF-α and IL-1β, respectively. Highly overlapping gene subsets were sensitive to inhibition by I-BET151 in both TNF-α and IL-1β-stimulated cells, representing gene products important in the initiation and perpetuation of inflammation and tissue destruction, such as cytokines, chemokines, Toll-like receptors (TLRs), MMPs and components of intracellular signalling cascades (figure 3A, B). Conversely, expression of genes involved in negative regulation of inflammatory activation (eg, NFKB1A, SOCS3), tissue-protective TIMP1 and antiproliferative (FoxO1, p21) genes were either unaffected or significantly increased (figure 3C, D). Control experiments demonstrated that effects of I-BET151 on RASF gene expression were dose-dependent, although dose sensitivity varied between gene targets (see online supplementary figure S2). Effects on gene expression could not be attributed to impairment of receptor-proximal signalling events, as I-BET151 had no effect on TNF-α and IL-1β-induced activation of mitogen-activated protein kinases, PKB or NF-κB pathways (see online supplementary figure S3A) as well as TNF-α and LPS-induced NF-κB luciferase activity in the absence of a natural chromatin structure (see online supplementary figure S3B). We also did not observe changes in the nuclear translocation and subsequent in vitro binding of NF-κB p50 and p65 subunits to immobilised oligonucleotides containing appropriate binding sites (see online supplementary figure S3C).

Effect of BET family inhibition on rheumatoid arthritis synovial fibroblasts (RASF) gene expression. RASF from three different RA patients were stimulated with TNF-α (A and C) or IL-1β (B and D) for 4 h in the presence or absence of 1 μM I-BET151. Total RNA was isolated, reverse transcribed and gene expression analysed by custom designed qPCR array. Genes in panels A–D were sorted according to mean suppression level observed in TNF-stimulated cells. Data are presented as per cent of mRNA expression in I-BET-treated cells as compared with controls incubated with cytokine alone. To compare differences in sensitivity to inhibition, genes were grouped into I-BET-inhibited (A and B) and I-BET unaffected/induced (C and D) subsets based on 25% inhibition cut-off after TNF stimulation. Gene subsets were analysed for overlap between both types of stimulation and functional role in inflammation. *p<0.05, **p<0.01, ***p<0.001.
I-BET151 suppresses the inflammatory activation of RASF by TLR ligands
As gene expression data indicated that I-BET151 significantly reduced expression of multiple TLRs, as well as their downstream signalling components, such as Myd88, we also examined if the compound could directly influence TLR-dependent gene expression in RASF. RASF were preincubated with I-BET151 prior to stimulation with the TLR ligands Pam3 (TLR2), pIC (TLR3) and LPS (TLR4), and mRNA expression and secreted products of MMP1, MMP3, IL-6 and IL-8 were assessed. At the mRNA level, regardless of the stimuli used, induction of all targets was significantly reduced in the presence of I-BET151 (figure 4A). At the protein level, I-BET151 only reduced the expression of MMP1 in response to pIC, but not other TLR ligands (figure 4B). However, I-BET151 significantly suppressed the production of MMP3, IL-6 and IL-8 (p<0.005–0.05) after stimulation with all TLR ligands tested. Together, these data indicate that inhibition of BET family proteins can differentially block the induction of gene expression by a wide variety of inflammatory stimuli present in RA synovial tissue.

Effect of I-BET151 on Toll-like receptor (TLR)-induced MMP and cytokine expression in rheumatoid arthritis synovial fibroblasts (RASF). RASF (n=12) were treated with the TLR ligands Pam3, pIC and LPS, or left unstimulated (control (ctrl)) for 24 h in the absence (ctrl) or presence of I-BET151 (I-BET), followed by measurement of MMP1, MMP3, IL-6 and IL-8 (A) mRNA expression and (B) protein concentration in cell-free tissue culture supernatants. Data in A are expressed as fold-mRNA expression compared with untreated, unstimulated RASF control cells. *p<0.05, **p<0.01, ***p<0.001.
Silencing of BET family proteins in RASF
We next attempted to independently confirm a role for BET family proteins in promoting the inflammatory activation of RASF. Since I-BET151 is an inhibitor of all three BET proteins,14 we performed triple transfections with siRNA targeting BRD2, BRD3 and BRD4 (siBRD2/3/4). Treatment of RASF with this siRNA cocktail resulted in 50%–60% reduction in BRD2 and BRD3 mRNA expression, and an approximately 80% reduction in BRD4 mRNA (figure 5A, top panel). Similar reductions in protein levels of each of the BET family proteins were also observed (figure 5A, bottom panel). In preliminary experiments, we detected no effect of transfected control siRNA on RASF gene expression compared with mock-transfected cells (see online supplementary figure S4). While there was no effect on basal or TNF-α-induced MMP1 production after silencing of BRD2/3/4, a partial reduction in IL-1β-induced MMP1 secretion was observed (p<0.01) (figure 5B). Similarly, siBRD2/3/4 had no effect on basal or cytokine-induced MMP3 and IL-8 secretion, and we observed a significant decrease in only basal, but not cytokine-induced, IL-6 production. We also examined the effects of siBRD2/3/4 on the mRNA expression of a selection of other gene targets we had identified as being modulated by I-BET151 (figure 5C). While cytokine-induced expression of CXCL9 was unaffected by triple transfection in contrast to treatment with I-BET151, siBRD2/3/4 suppressed TNF-α-induced CXCL10 and CXCL11 expression, and enhanced TIMP1 expression in response to both TNF-α and IL-1β, similar to what was observed with I-BET151. Examining the effects of siBRD2/3/4 in TLR-stimulated RASF, some overlap with the effects of I-BET151 was also observed (see online supplementary figure S5). Together, these experiments indicate that partial silencing of BET family proteins in RASF mimics a subset but not all of the broad anti-inflammatory effects of I-BET151. To independently validate the specificity of I-BET151, we compared gene expression patterns in RASF stimulated with IL-1β in the presence of I-BET151 and another BET protein inhibitor, JQ1 (figure 5D, and see online supplementary figure S6). Patterns observed for both inhibitors were nearly identical (r=0.9899, p<0.0001), strongly supporting I-BET151 mechanism of action as being reliant on BET protein inhibition.

Effects of BET family protein silencing on gene expression in rheumatoid arthritis synovial fibroblasts (RASF). (A) RASF were transfected with a pool of siRNAs targeting BRD2, BRD3, and BRD4 or scrambled control (ctrl) siRNA and BRD2, BRD3 and BRD4 mRNA quantified relative to ctrl siRNA-treated cells (n=9) (top panel). Alternatively, BRD2, BRD3 and BRD4 were detected by Western blotting (bottom panel). (B and C) RASF were treated as in (A) and left unstimulated (medium) or stimulated with TNF-α or IL-1β for 24 h. (C) Tissue culture supernatant concentrations of MMP1, MMP3, IL-6 and IL-8 were measured by ELISA (n=8). (D) CXCL9, CXCL10, CXCL11 and TIMP1 mRNA were assessed by q-PCR (n=9). (D) RASF (n=3) were left untreated or preincubated with 1 µM I-BET151 or 1 µM JQ1 for 1 h and subsequently stimulated with 1 ng/mL IL-1β for 4 h. Total RNA was extracted, reverse transcribed and gene expression analysed by low-density qPCR gene arrays. Fold changes in expression of 84 genes between cells stimulated with IL-1β in the presence or absence of each inhibitor were log transformed and correlation between mean values for I-BET and JQ1 analysed by Pearson correlation. *p<0.05, **p<0.01, ***p<0.001.
I-BET151 suppresses the proliferation and chemo-attractant abilities of RASF
Next, we asked whether the changes we observed in expression levels of genes regulating cell survival, proliferation and chemotactic capacity translated into differences in the functional behaviour of RASF. In initial experiments, we noted no effects of I-BET151 on RASF mitochondrial viability either in unstimulated or cytokine-stimulated RASF (figure 6A). Additionally, I-BET151 by itself or in cooperation with TNF-α or IL-1β failed to induce RASF apoptosis (figure 6B). However, I-BET151 reduced the proliferation rate of RASF (figure 6C) in medium alone (66.9±12.1%) as well as following stimulation with TNF-α (69.6±13.8%), IL-1β (71.0±15.0%), Pam3 (60.0±20.0%) and LPS (68.0±18.0%) (p<0.01 for all conditions). In contrast, I-BET151 failed to reduce pIC-induced proliferation rates. To examine potential effects of I-BET151 on the ability of RASF to recruit immune cells, we treated RASF overnight in the absence or presence of I-BET151 in medium alone or in combination with inflammatory stimuli. Cell-free supernatants were collected and used as a chemotactic source for healthy donor peripheral blood leucocytes. As expected, based on gene expression data, I-BET151 significantly reduced RASF chemo-attractant potential in response to TNF-α, and pIC and LPS, and similar trends were observed in RASF stimulated by IL-1β or Pam3 (figure 6D). Together, these data indicate that changes in RASF gene expression following I-BET151 exposure are sufficient to modulate the proliferative and chemo-attractant potential of RASF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of I-BET151 on rheumatoid arthritis synovial fibroblasts (RASF) functional properties. (A) RASF were left untreated or stimulated with TNF-α and IL-1β for 24 h in the presence or absence of indicated doses of I-BET151. (A) Metabolic activity was assessed by MTT assay (n=3–4) and (B) apoptosis induction by cell death detection ELISA assay (n=3). (C) Proliferation of RASF (n=9) in presence and absence of 1 μM I-BET151 and TNF-α, IL-1β or the Toll-like receptor (TLR) ligands Pam3, pIC and LPS was measured by colorimetric cell proliferation ELISA. Proliferation of I-BET151-treated RASF is shown relative to untreated RASF (normalised to one). (D) RASF (n=5) were incubated in presence or absence of 1 μM I-BET151 and stimulated with TNF-α, IL-1β or the TLR ligands Pam3, pIC or LPS for 24 h. Peripheral blood leucocytes from healthy donors were incubated with conditioned medium from RASF in a transwell system for 6 h and the number of migrating leucocytes was counted. *p<0.05, **p<0.01.
Discussion
Our data provide the first evidence suggesting a therapeutic potential for targeting epigenetic reader proteins in RA, showing that I-BET151 suppresses TNF-α, IL-1β and TLR ligand-induced expression of key molecules in RASF that contribute to pathology, including MMPs, cytokines and chemokines. These changes in expression levels translated directly into functional consequences for RASF cellular behaviour, including reduced proliferation rates and diminished capacity to recruit immune cells. Interestingly, two previous studies had provided indirect evidence indicating a potential role for BET bromodomain proteins in the pathology of RA. Three single nucleotide polymorphisms (SNPs) in the BRD2 locus have been identified which are associated with an increased risk for RA.24 Additionally, Brd2 was identified as a gene that is differentially expressed in OA and RA SF, although at the time, the role of Brd2 in transcriptional regulation was not yet appreciated.25 A SNP in another, non-BET bromodomain protein, BRD1, was shown to be associated with joint destruction in RA in stage I of a genome-wide association study.26 In light of our evidence that pharmacological inhibition of BET family proteins suppresses inflammatory activation of RASF, it will be of interest to determine the potential contributions of SNPs in loci encoding bromodomain proteins to the regulation of gene transcription in RA.
In attempts to validate the specificity of effects of I-BET151 on RASF gene expression, we were able to obtain only a partial silencing of BRD2/3/4. Despite this, we observed some similar effects on a subset of I-BET151-regulated genes tested, including MMP1, TIMP1, CXCL10 and CXCL11, but not on IL-6 and IL-8. Although, based on these studies, we cannot rule out off-target effects of I-BET151 as such, comparison between I-BET151 and JQ1, another thoroughly described and specific but chemically unrelated inhibitor of BET proteins, strengthens the idea that I-BET151 mediates its anti-inflammatory effects via BET family proteins. Extensive screening of the prototypical compound I-BET762 has failed to identify non-BET targets, and similar effects of I-BET151 and I-BET762 were observed in functional assays and proteomic profiling.14 ,27 The relative reliance of inflammatory gene expression on BET family members could vary between cell types, as in murine bone marrow-derived macrophages expressing a hypomorphic Brd2, resulting in a 50% decrease in protein expression, decreases in cytokine and chemokine production closely mimic effects of the BET inhibitor JQ-1.15 A similar overlap in effects on gene expression was also observed when murine macrophages were either treated with I-BET762 or subjected to BRD2/3/4 silencing treatment.14 However, some differences are observed, as macrophage expression of TNF-α was resistant to I-BET762 treatment, while silencing of BET proteins in macrophages suppressed the LPS-induced expression of TNF-α.14 This may suggest the existence of BET-recruiting mechanisms that are independent of BET interaction with acetylated histones or an effect of sustained protein/complex loss incurred from extended siRNA treatment compared with acute exposure to I-BET151.14 BRD2 has been shown to interact with proteins exhibiting transcriptional repressor function and act as a scaffolding protein for a coordinated positive and negative regulation of gene promoters,13 and BRD4 has been demonstrated to have chromatin-independent properties in transcriptional activation by binding to acetylated RelA and serving as a cofactor for NF-κB activation,28 a mechanism apparently not operational in RASF based on our analyses of NF-κB transcriptional and DNA binding activity. Thus, yet unidentified and likely cell type-specific mechanisms are probably involved in the I-BET151-mediated effects on some aspects of gene expression regulation.
Initial characterisations of I-BET compounds, as well as JQ-1, have demonstrated functional roles for BET proteins in controlling acute and chronic inflammatory responses, both in vitro and in vivo. In these studies, anti-inflammatory activity was observed on macrophages and Th1 and Th17 T lymphocytes, all cell populations which contribute to pathology in RA and other forms of inflammatory arthritis.29–32 I-BET762 can disrupt chromatin complexes responsible for the expression of key inflammatory genes in LPS-activated macrophages and has proven protective in bacteria-induced sepsis mouse models.14 Additionally, treatment of naive CD4+ T cells with BET inhibitors suppresses the pro-inflammatory functions of Th1 cells as well as differentiation towards and activation of previously differentiated Th17 cells, and protects mice in models of multiple sclerosis and arthritis.16 ,17 These observations, together with results from our study, suggest that specific targeting of epigenetic reader proteins, in particular BET family members, may have therapeutic potential in blocking the inflammatory activation of multiple cell types important in pathology in RA and other immune-mediated inflammatory diseases.
Acknowledgments
We thank Maria Comazzi, Peter Künzler, Desiree Pots and Inge van Es for excellent technical assistance. We thank Professor Dr Beat A Michel for his support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table
Footnotes
Handling editor Tore K Kvien
KK, PAK, CO and KAR contributed equally.
Contributors KK, PAK and AMG contributed to research design, performed experiments, analysed data and contributed to writing the paper; CK obtained, recorded, analysed and interpreted all clinical records and materials; REG, L-LL, SG, PPT, RKP, CO and KAR designed research, analysed and interpreted data, and contributed to writing the manuscript.
Funding The research leading to these results has received funding from the Innovative Medicines Initiative (IMI) BTCure (grant number 115142), EuroTEAM (grant number 305549), the Institute of Arthritis Research (IAR), Epalinges and GlaxoSmithKline.
Competing interests This research was supported by an open research programme grant from GlaxoSmithKline to KAR. L-LL is an employee and shareholder of Pfizer. PPT and RKP are employees and shareholders of GlaxoSmithKline.
Ethics approval Swiss Ethical Commission and Commission of the Academic Medical Center, University of Amsterdam, Amsterdam.
Provenance and peer review Not commissioned; externally peer reviewed.