Article Text

Abstract
Objectives Patients with antiphospholipid syndrome (APS) are at risk for subclinical endothelial injury, as well as accelerated atherosclerosis. In the related disease systemic lupus erythematosus, there is a well-established defect in circulating endothelial progenitors, which leads to an accrual of endothelial damage over time. This defect has been at least partially attributed to exaggerated expression of type I interferons (IFNs). We sought to determine whether these pathways are important in APS.
Methods We studied 68 patients with primary APS. Endothelial progenitors were assessed by flow cytometry and functional assay. Type I IFN activity was determined by a well-accepted bioassay, while peripheral blood mononuclear cells were scored for expression of IFN-responsive genes.
Results Endothelial progenitors from patients with APS demonstrated a marked defect in the ability to differentiate into endothelial cells, a phenotype which could be mimicked by treating control progenitors with APS sera. Elevated type I IFN activity was detected in the circulation of patients with APS (a finding that was then replicated in an independent cohort). While IgG depletion from APS sera did not rescue endothelial progenitor function, the dysfunction was successfully reversed by a type I IFN receptor-neutralising antibody.
Conclusions We describe, for the first time to our knowledge, an IFN signature in primary APS and show that this promotes impaired endothelial progenitor function. This work opens the door to novel approaches that may mitigate vascular damage in APS, such as anti-IFN drugs.
- Antiphospholipid Syndrome
- Atherosclerosis
- Cytokines
Statistics from Altmetric.com
Introduction
Endothelial dysfunction and preclinical endothelial damage are well-recognised complications of diseases such as diabetes, rheumatoid arthritis and systemic lupus erythematosus (SLE). Not coincidentally, these diseases are also strongly associated with accelerated atherosclerosis and significant cardiovascular morbidity. Indeed, endothelial dysfunction and damage serve as predictors, and perhaps precipitators, of atherosclerotic lesions.
These preclinical endothelial abnormalities are at least partially attributable to low numbers and function of so-called endothelial progenitors.1–3 Endothelial progenitors are a heterogeneous collection of circulating cells with proangiogenic functions including the production of cytokines like vascular endothelial growth factor (VEGF),4 and the ability to home to ischaemic tissue where they support angiogenesis.5 Some subtypes of progenitors also likely differentiate into true endothelial cells (ECs) in vivo, although this concept is somewhat controversial and continues to be explored.6 ,7
The first endothelial progenitor to be described was the classic endothelial progenitor cell (EPC). Based on surface markers, EPCs are believed to originate from the hemangioblast, the pluripotent bone marrow progenitor of both haematopoietic cells and ECs. EPCs are typically defined and quantified in peripheral blood based on the surface profile CD34+CD133+VEGFR2+.8 ,9 Another strategy for characterising progenitors does not rely on surface markers, but rather the ability of cells from the peripheral blood mononuclear cell (PBMC) fraction to take on features of mature ECs over days to weeks when cultured under well-defined proangiogenic conditions.1 ,10 At the end of this period, EC-like cells are identified by their ability to take up low-density lipoprotein and bind specified lectins. While some of these cells are derived from the aforementioned EPCs, many have the surface profile CD14+CD45+, and are presumed to originate from the myeloid compartment.4 These myeloid-derived cells were originally named circulating angiogenic cells (CACs),4 which is the nomenclature that will be used in this report. Other names have also been applied in the literature including myeloid EPCs, early EPCs and proangiogenic cells (PACs).6 ,7 Importantly, CACs, similar to classic EPCs, have potent proangiogenic properties.4 ,11
Antiphospholipid syndrome (APS) is a leading acquired cause of both thrombophilia and pregnancy loss. Approximately 50% of cases occur in patients with systemic lupus, while the remaining diagnoses comprise a standalone syndrome referred to as primary APS.12 APS is characterised by the presence of so-called antiphospholipid antibodies. This historical name belies the fact that pathogenic antibodies in APS do not actually target phospholipids, but rather clotting factors and other lipid-binding proteins. Indeed, the best-described antigenic target in APS is a protein that circulates at high levels in blood called β-2 glycoprotein I (β2GPI).13 ,14 Work over the past three decades has explored the ability of anti-β2GPI antibodies to stimulate the endothelium and platelets,15 while more recent studies have also implicated activation of neutrophils and neutrophil extracellular traps (NETs) in APS pathogenesis.16 ,17
In addition to its cardinal manifestations of thrombosis and pregnancy loss, APS is also characterised by seizures, cognitive decline, thrombocytopenia and nephropathy, highlighting its status as a truly systemic autoimmune disease.18 Further, accelerated atherosclerosis (which is not explained by traditional cardiovascular risk factors) is another recognised complication of APS.19 ,20 As presumed precursors to atherosclerosis, subclinical endothelial damage and dysfunction have been described in many,21–25 but not all23 ,26 studies of primary APS. Indeed, a recent meta-analysis of APS case-control studies found evidence of both increased carotid intima-media thickness and decreased flow mediated dilation,27 both of which are indicative of endothelial injury.
The status of endothelial progenitors has only been investigated to a limited extent in APS. Gresele and colleagues characterised 20 cases of primary APS, and in particular focused on patients with no traditional cardiovascular risk factors.26 As compared with matched controls, they found no difference in the levels of EPCs (CD34+CD133+VEGFR2+) in circulation.26 Further, in a subgroup analysis of an SLE study, Denny and colleagues found no influence of anticardiolipin, anti-β2GPI or lupus anticoagulant on circulating EPC numbers (CD34+CD133+).1
As mentioned above, there is a well-established reduction in the numbers and function of circulating endothelial progenitors in SLE, which potentially leads to an accrual of endothelial damage over time. This defect is at least partially attributable to elevated levels of type I interferons (IFNs).28 Indeed, type I IFN expression correlates strongly with subclinical vascular disease,29 ,30 as well as abnormal numbers and function of endothelial progenitors.1 ,30 Endothelial progenitor defects in SLE have been observed both by flow cytometry of peripheral blood for CD34+CD133+ EPCs,1 and by functional assays that simultaneously characterise both EPCs and CACs.29 ,30 Further, IFN-associated endothelial progenitor dysfunction has been reported in rheumatoid arthritis,31 paediatric lupus32 and murine models of lupus.33 ,34 Mechanistically, IFN-α (the type I IFN most closely aligned with SLE) represses IL-1 signalling and thereby skews myeloid cells away from the CAC phenotype.35 IFN-α has also been reported to function through the inflammasome to activate IL-18, a cytokine that turns out to be deleterious to CAC differentiation.10 Of note, the experiments that mechanistically implicate type I IFNs in the endothelial progenitor dysfunction of SLE have primarily involved functional assays that simultaneously report on both EPCs and CACs. In this study, we set out to explore endothelial progenitor function in primary APS, with those findings leading us to also characterise type I IFN expression.
Methods
See online supplementary material methods for further details.
Supplemental material
Human subjects
None of the 42 patients with primary APS met American College of Rheumatology criteria for SLE.36
In vitro differentiation into mature ECs
EC-enrichment culture media was prepared by supplementing MCDB 131 (Gibco) with endothelial cell growth medium (EGM) SingleQuot factors and 20% heat-inactivated fetal bovine serum. In some cases, the media was additionally supplemented with 30% patient or control sera. PBMCs were seeded into 48-well Human Fibronectin Cellware. After 12–14 days, cells were incubated with markers of mature ECs, specifically fluorescein-conjugated Ulex europaeus agglutinin-1 lectin and DiI-labelled acetylated low-density lipoprotein (LDL). In some experiments, a neutralising monoclonal antibody against the type I IFN receptor was included in the media during the first 3 days of culture.
HeLa cell cultures
Cells were cultured in Eagle’s minimal essential medium (EMEM) media (Lonza), which was supplemented with 50% patient or control sera.
Results
While it is recognised that patients with primary APS are susceptible to endothelial dysfunction and damage,27 the status of their endothelial progenitors has not been thoroughly investigated. In this study, we characterised the blood of 42 patients with primary APS and 63 non-autoimmune controls as our primary cohort (table 1). The patients and controls did not differ significantly for age or gender, and were always matched for these factors within individual experiments. The patients with APS had antibody profiles and clinical manifestations as described in table 1.
Patients with APS and controls
Patients with APS have a reduction in functional EPC/CACs
We first assessed classic EPCs in patients with APS as compared with controls. Like others, we defined these cells as CD34+CD133+.1 ,32 Although there was a trend towards decreased EPC numbers in primary APS, this did not reach statistical significance (figure 1A). We then turned our attention to a functional assay that characterises a broader set of endothelial progenitors, including CACs (which are derived from the myeloid compartment) and EPCs.1 ,4 Because this assay characterises the PBMC fraction for both EPCs and CACs, we will subsequently refer to progenitors scored by this assay as EPC/CACs. Indeed, PBMC fractions from patients with primary APS displayed a striking reduction in the number of EPC/CACs that successfully differentiated into EC-like cells (figure 1B, C). These results suggest that while there is no significant reduction in classic EPCs in primary APS, a broader look at endothelial progenitors does reveal a significant defect in patients as compared with controls.
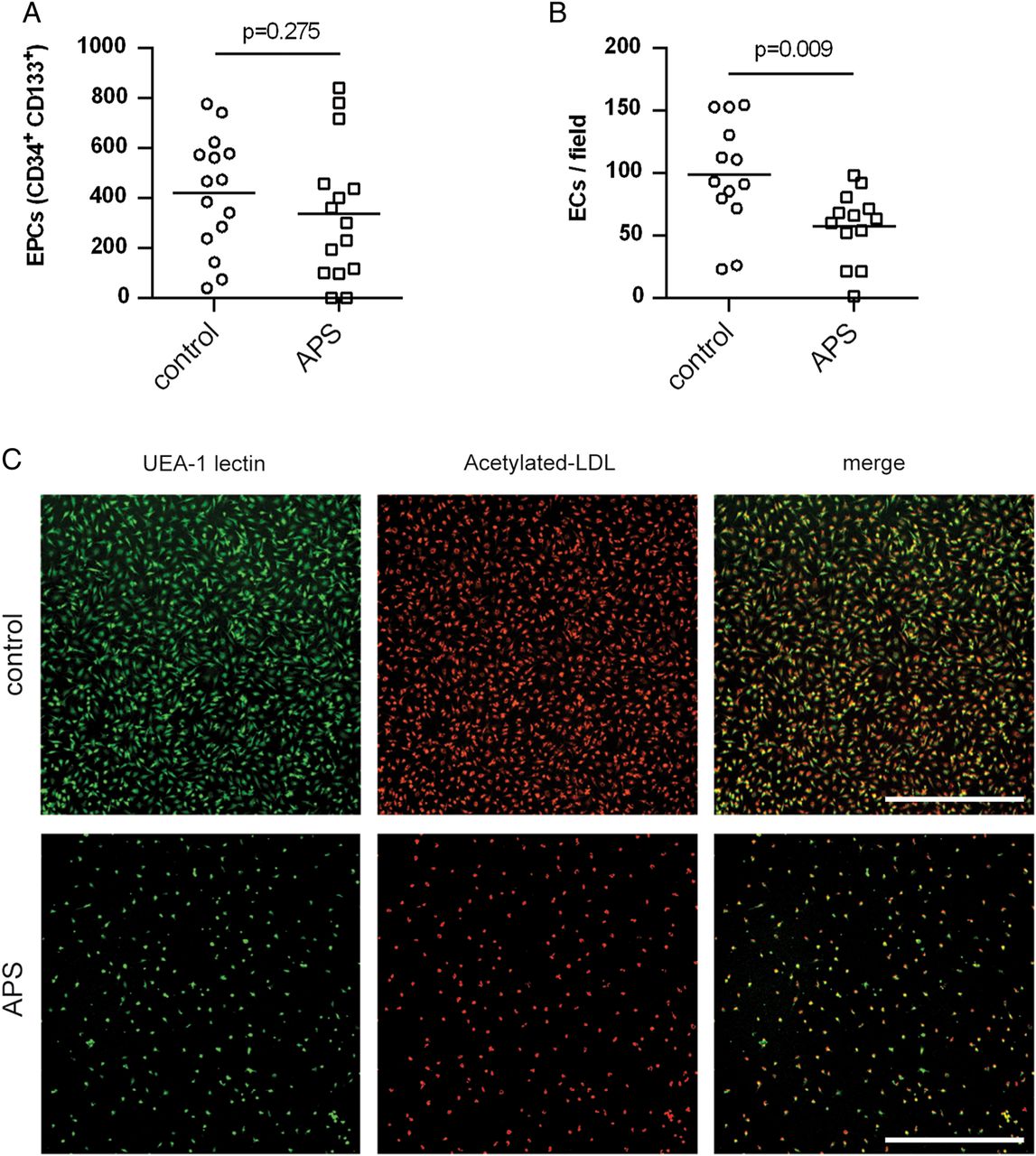
Endothelial progenitor cell (EPC)/circulating angiogenic cells (CACs) show a diminished ability to differentiate into endothelial cell (EC)-like cells in primary antiphospholipid syndrome (APS). (A) Classic EPCs (defined as CD34+CD133+) were quantified in peripheral blood of controls and patients with primary APS by flow cytometry (units=EPCs/mL of blood). Each data point represents an individual control/patient. The mean for each group is represented by a horizontal line. (B) Peripheral blood mononuclear cells were isolated from either controls or patients with primary APS, and then cultured under proangiogenic conditions for 12–14 days. EPC/CACs were scored for their ability to successfully differentiate into EC-like cells as determined by surface binding of both Ulex europaeus agglutinin (UEA)-1 lectin and acetylated LDL. Again, means and individual data points are indicated. (C) Representative photomicrographs from the experiments of B. UEA-1 lectin is stained green and acetylated LDL red. Scale bar=500 µm.
APS sera impair the differentiation of control EPC/CACs into EC-like cells
We next tested whether the impairment of EPC/CACs observed in APS might be attributable to factors that PBMCs were exposed to in circulation. Specifically, we cultured control PBMCs with heterologous control or APS sera. We found that the APS sera (as compared with control sera) significantly reduced the number of EPC/CACs successfully differentiating into EC-like cells (figure 2A, B). We asked whether APS IgG might directly impair the differentiation of EPC/CACs into EC-like cells, and found that depletion of total IgG from APS sera did not rescue EPC/CAC differentiation (figure 2C, D). We also cultured control PBMCs in the presence of IgG purified from heterologous control or APS sera. Corroborating the depletion experiment, purified APS IgG did not interfere with EPC/CAC differentiation (see online supplementary figure S1). In summary, these data suggest that there are factors in APS sera (other than the antiphospholipid antibodies themselves) that impair successful differentiation of EPC/CACs into EC-like cells.

Primary antiphospholipid syndrome (APS) sera interfere with the emergence of endothelial cell (EC)-like cells from control endothelial progenitor cell (EPC)/circulating angiogenic cells (CACs). (A) Peripheral blood mononuclear cells (PBMCs) were isolated from controls, and then cultured under proangiogenic conditions for 12–14 days. For the first 3 days of culture, media was supplemented with sera from either heterologous controls or patients with primary APS. EPC/CACs were scored for their ability to successfully differentiate into EC-like cells as determined by surface binding of both Ulex europaeus agglutinin (UEA)-1 lectin and acetylated LDL. Each data point represents an individual control/patient. The mean for each group is represented by a horizontal line. (B) Representative photomicrographs from the experiments of A. UEA-1 lectin is stained green and acetylated LDL red. Scale bar=500 µm. (C) Total IgG was depleted from 10 APS serum samples. These patients were all positive for anti-β2 glycoprotein I (GPI) IgG at baseline. For each group, the median, 25th/75th percentiles and minimum/maximum are represented by box and whisker. Anti-β2GPI IgG was undetectable in the depleted samples. (D) As in A, PBMCs were isolated from controls, and then cultured under proangiogenic conditions for 12–14 days. For the first 3 days of culture, media was supplemented with APS sera that were either depleted of IgG, or not. EPC/CACs were scored for their ability to successfully differentiate into EC-like cells as determined by surface binding of both UEA-1 lectin and acetylated LDL. Each pair of data points represents an individual patient.
APS sera have elevated levels of type I IFNs
As discussed above, SLE is associated with a well-known defect in numbers and function of endothelial progenitors, a phenotype that is at least partially attributable to expression of type I IFNs. Further, the impact of type I IFNs has been especially demonstrable in functional experiments requiring EPC/CACs to differentiate into EC-like cells, such as the assay used here (figure 2). We therefore explored whether sera from patients with APS might have evidence of exuberant type I IFN activity. To detect this activity, we used a well-accepted bioassay29 and quantitation of three IFN-responsive genes judged to be relatively specific for type I IFNs as compared with type II IFNs such as IFN-γ.37 Advantages of the bioassay include its avoidance of the ELISAs that have not always proven reliable or sensitive for IFN-α,38 and also its ability to capture data on other biologically active type I IFNs in addition to IFN-α. Interestingly, we found that APS sera strongly induce the expression of all three IFN-responsive genes in HeLa cells (as compared with heterologous control sera), consistent with the presence of higher type I IFN activity in APS (figure 3). While HeLa cells have been used previously for the bioassay,29 we also sought confirmation in cells that might have more relevance for EPC/CAC differentiation, namely human umbilical vein endothelial cells (HUVECs). Indeed, APS sera induced the expression of IFN-responsive genes in HUVECs (see online supplementary figure S2). We further went beyond the bioassays by testing for IFN-α directly in sera with a commercial ELISA that is designed to capture all relevant IFN-α subtypes. Again, we revealed upregulation of IFN-α in APS sera as compared with control (see online supplementary figure S3).

Primary antiphospholipid syndrome (APS) sera have elevated levels of type I interferons (IFNs) based on an established bioassay. As described in Methods, HeLa cells were treated with sera from either controls or patients with primary APS for 6 hours, and then harvested for RNA preparation. Expression of the type I IFN-regulated genes IFIT1 (A), IFI44 (B) and PRKR (C) was scored by quantitative PCR. Each data point represents an individual control/patient. The median for each group is represented by a horizontal line.
To our knowledge, this is the first report of increased IFN activity in primary APS. We therefore tested serum samples from an independent cohort of 26 patients with primary APS, recruited from a different centre (see online supplementary table S1). As compared with 26 non-autoimmune controls, the patients with primary APS again showed robust IFN upregulation, as determined by the HeLa bioassay (see online supplementary figure S4). In summary, these data demonstrate evidence of elevated type I IFN activity in primary APS, spanning two independent patient cohorts.
APS PBMCs display an IFN signature
Having demonstrated elevated type I IFNs in APS sera, we asked whether there was evidence that APS PBMCs had been exposed to higher-than-normal IFN activity in vivo. This is a relevant question as PBMCs from patients with primary APS differentiated less efficiently into EC-like cells in the experiments of figure 1. Indeed, primary APS PBMCs (similar to SLE PBMCs) showed upregulation of three IFN-responsive genes (figure 4A–C and online supplementary table S2). Again, these are three genes with relative specificity for type I IFNs as compared with IFN-γ.37 When we segregated the patients according to whether they had positive testing for anti-β2GPI IgG by the Sydney criteria, we found that PBMCs from the anti-β2GPI IgG-positive patients demonstrated higher levels of two of the three IFN-responsive genes, with a trend in the same direction for the third gene (figure 4D). We additionally segregated patients by the other clinical manifestations and lab testing that are part of the Sydney criteria, but did not find other statistically significant correlations (see online supplementary figures S5 and S6). In summary, these experiments suggest that in APS the fraction of cells containing EPC/CACs (ie, PBMCs) has evidence of elevated IFN exposure in vivo.
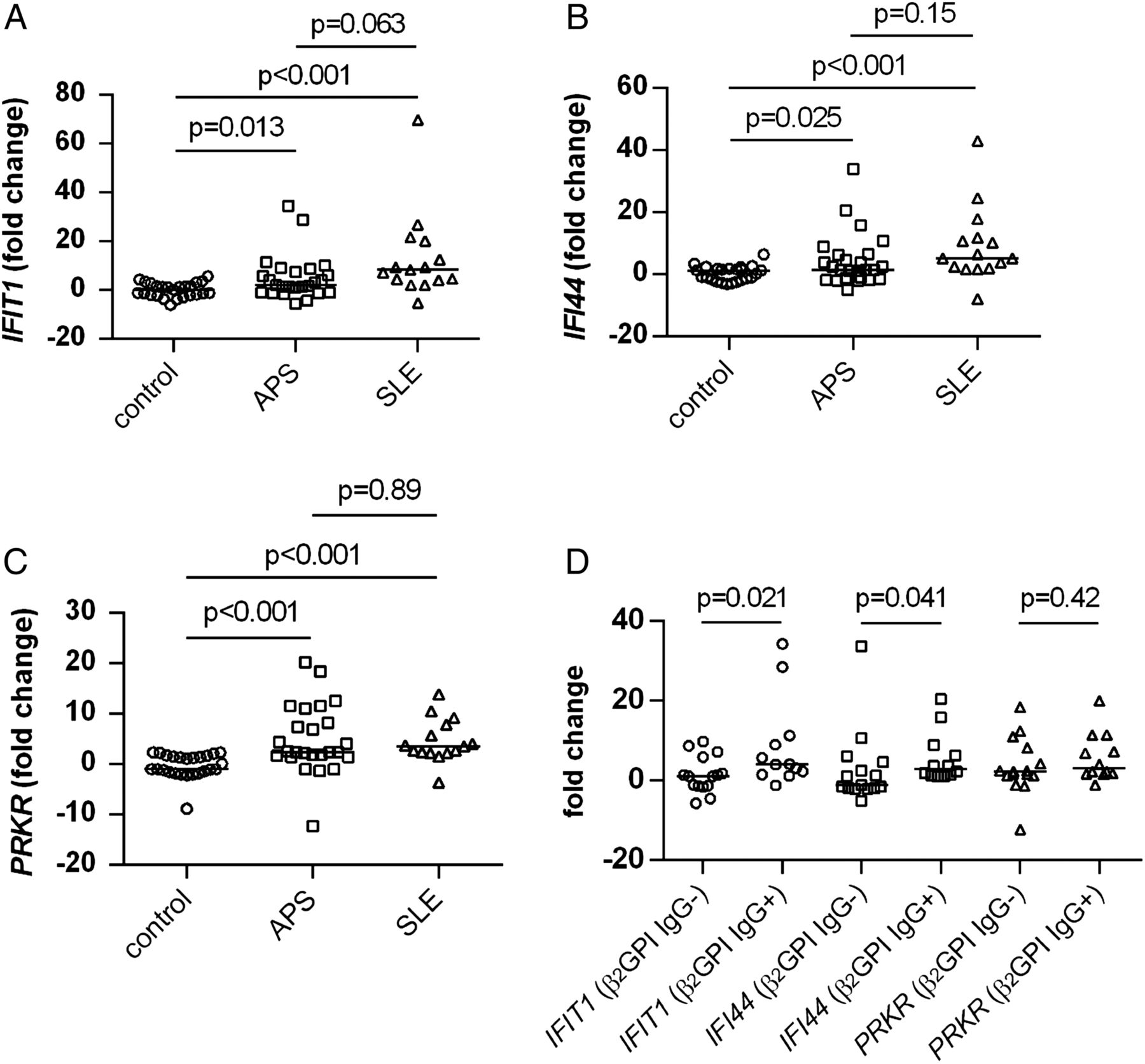
The peripheral blood mononuclear cell (PBMC) compartment has evidence of a type I interferon (IFN) signature in patients with primary antiphospholipid syndrome (APS), similar to patients with systemic lupus erythematosus (SLE). RNA was prepared from total PBMCs. Expression of the type I IFN-regulated genes IFIT1 (A), IFI44 (B) and PRKR (C) was scored by quantitative PCR. Each data point represents an individual control/patient. The median for each group is represented by a horizontal line. (D) Data for patients with primary APS are segregated based on whether the patients were negative or positive for anti-β2 glycoprotein I (GPI) IgG by the Sydney criteria. Again, medians and individual data points are indicated.
Blockade of the type I IFN receptor rescues the differentiation of EPC/CACs into EC-like cells
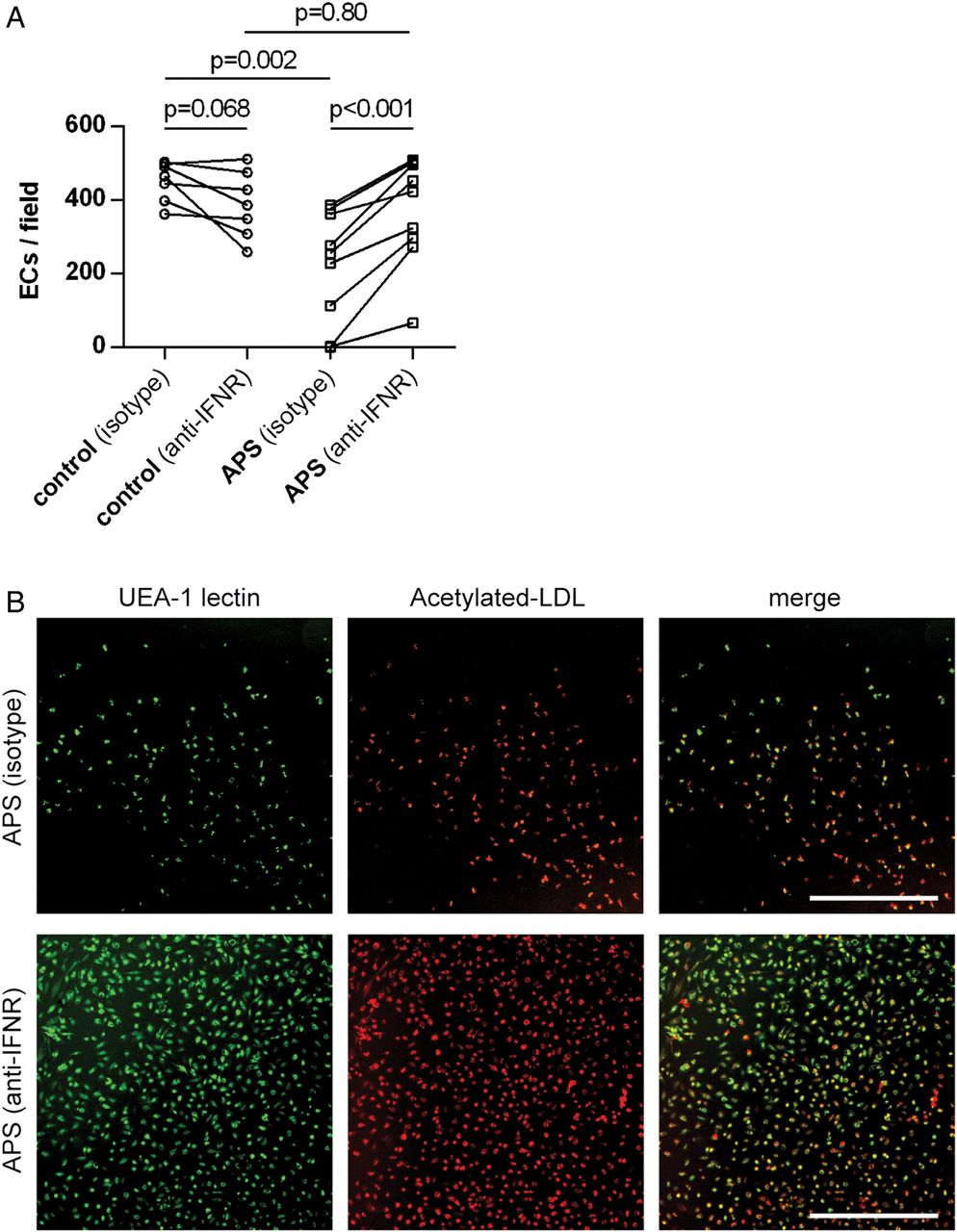
Finally, we revisited the experiments of figure 2, in which APS sera interfered with the differentiation of control EPC/CACs into EC-like cells. Specifically, we tested 10 APS sera (all patients were positive for anti-β2GPI IgG) in the presence or absence of a monoclonal antibody that neutralises the type I IFN receptor. While the antibody had no effect in the context of control sera (figure 5A), it rescued EPC/CAC differentiation when PBMCs were exposed to APS sera (figure 5A, B). This result suggests that an important means by which APS sera reduce functional EPC/CAC differentiation is through type I IFN signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A neutralising antibody against the type I interferon receptor (IFNR) restores normal endothelial progenitor cell (EPC)/circulating angiogenic cell (CAC) differentiation in patients with primary antiphospholipid syndrome (APS). (A) Peripheral blood mononuclear cells were isolated from controls, and then cultured under proangiogenic conditions for 12–14 days. For the first 3 days of culture, media was supplemented with sera from either heterologous controls or patients with primary APS. For these same days, media was additionally supplemented with the aforementioned IFNR-neutralising antibody, or isotype control. EPC/CACs were scored for their ability to successfully differentiate into EC-like cells as determined by surface binding of both Ulex europaeus agglutinin (UEA)-1 lectin and acetylated LDL. Each pair of data points represents an individual control/patient. (B) Representative photomicrographs from the experiments of A. UEA-1 lectin is stained green and acetylated LDL red. Scale bar=500 µm.
Discussion
Primary APS is associated with endothelial dysfunction and damage, and we now show that this may be at least partially attributable to a defect in endothelial progenitors.27 Interestingly, the numbers of circulating classic EPCs were not appreciably different between primary APS and controls, which is in line with the work of another group.26 However, when we looked more broadly at endothelial progenitors using a well-established functional assay, we found that patients with primary APS have a clear reduction in numbers of functional EPC/CACs. After observing this phenotype in primary cells from patients with APS, we also demonstrated that primary APS sera impair the ability of control EPC/CACs to take on features of EC-like cells in the functional assay. We considered the possibility that this might be attributable to the antiphospholipid antibodies themselves, but IgG depletion did not rescue EPC/CAC differentiation. Rather, we now favour the idea that circulating type I IFNs are a critical factor in preventing EPC/CAC maturation. Indeed, we found elevated IFNs in patient sera (which was replicated in an independent cohort), and an IFN signature in the PBMC fractions themselves. Importantly, we also showed that a neutralising antibody against the type I IFN receptor leads to a restoration of normal EPC/CAC differentiation.
This work mirrors studies in SLE, where it has been suggested that type I IFNs tip myeloid-lineage cells away from the role of endothelial progenitors and towards antigen presentation (eg, as dendritic cells).28 Although this is the first study that, to our knowledge, explicitly explores type I IFN expression in large primary APS cohorts, there are certainly other hints in the literature regarding IFN dysregulation in APS. For example, the expression of MCP1 and IFIT1 (two well-established IFN-regulated genes) correlates with anticardiolipin antibody levels in patients with SLE with secondary APS.39 Similarly, a pilot study of fluvastatin in antiphospholipid antibody-positive patients also suggested higher IFN-α levels by an ELISA assay.40 Another very small study that included two patients with primary APS described a type I IFN signature in those patients.41
The source and driver of type I IFNs in primary APS remains to be elucidated. We considered the possibility that our cohort of patients with primary APS may uniquely have more lupus features than most. This is argued against by the identification of elevated IFN activity in a completely independent cohort of patients with primary APS. Further, of the 42 patients with primary APS from our original cohort, 20 had a positive anti-nuclear antibody (ANA) by HEp-2 immunofluorescence, most at a titre of 1:160 or less. This is in line with other studies, which have found ANA prevalence on the order of 30%–40% in primary APS.42 ,43 Eleven patients had detectable anti-dsDNA, but all at low titres; no patient had detectable anti-Sm. Further, no patient with APS had a history of recurrent hypocomplementaemia, nor was any patient hypocomplementemic at the time of the donation. Also, no patient designated as having primary APS was being treated by their haematologist or rheumatologist for SLE according to clinical documentation. As type I IFN activity has previously been shown to differentiate asymptomatic autoantibody-positive patients from those with at least some features of SLE, one wonders if that might also be true for our cohort.44 We did not identify such features by retrospective chart review, but cannot rule out that they may develop going forward.
A related question is why previous studies focused on patients with SLE have not seen a correlation between presence of antiphospholipid antibodies and IFN production.45 We would point out that all of our patients meet criteria for APS, meaning their antibodies are durably positive, and also associated with thrombotic events or pregnancy morbidity. Further, within our population of patients with APS, the best lab predictor of high IFN expression was anti-β2GPI, which was not considered in the most comprehensive prior work.45
It is possible that toll-like receptors (TLRs) may play a role in driving type I IFN production in APS (as has been well described in SLE). For example, an interesting study has demonstrated that antiphospholipid antibodies promote the translocation of TLR7/8 to the endosome in monocytes and plasmacytoid dendritic cells, where they may then initiate IFN signalling. Further, work from our group has shown that NETs circulate at higher levels in primary APS, as compared with controls;16 indeed, NETs have been shown to activate TLRs and type I IFN production in SLE.46 ,47 It is also worth noting that the best-described spontaneous mouse model of APS (NZWx BXSB F1) overexpresses TLR7.48 One might even speculate about a possible role of type I IFNs in promoting antiphospholipid antibody formation given numerous reports of iatrogenic APS in patients treated with IFN-α for hepatitis C and melanoma.49–51
A limitation of our study is that while our patient cohorts are well-characterised for standard clinical variables, we do not have data regarding endothelial function/dysfunction for the patients recruited into this study. Follow-up work will explore the EPC/CAC defect, as well as type I IFN expression, in a cohort where such data are available. It will also be of interest to explore the impact of type I IFNs on mature ECs in APS,52 ,53 and to consider anti-type I IFN therapeutics in both human and murine APS—a concept that has been met with mixed success to this point in SLE.54 ,55 Indeed, better and more targeted therapeutics for APS are clearly needed as both criteria (thrombosis, pregnancy loss) and non-criteria (accelerated atherosclerosis, cognitive decline) manifestations of APS frequently progress despite current standard-of-care treatment.
Acknowledgments
The authors thank Emily Lewis and Emily Siegwald for assistance with patient recruitment.
References
Footnotes
Handling editor Tore K Kvien
RCG and SY contributed equally.
Twitter Follow Jason Knight at @jasonsknight
Contributors RCG, SY, AAG, NMK, CN-A and DH-R conducted experiments and analysed data. ARC, WJM, PLB and JSK analysed data and designed the study. All authors participated in writing the manuscript, and gave approval before submission.
Funding JSK was supported by NIH K08AR066569 and a career development award from the Burroughs Wellcome Fund. NMK received funding from the Security Forces Hospital Program, Ministry of Interior, Riyadh, Saudi Arabia. WJM and the Michigan Lupus Cohort were supported by the Mary Piazza Lupus Research Fund and the Michael and Marcia Klein Lupus Research Fund.
Competing interests None declared.
Ethics approval University Institutional Review Boards.
Provenance and peer review Not commissioned; externally peer reviewed.