Article Text

Abstract
Objectives MAXIMISE (Managing AXIal Manifestations in psorIatic arthritis with SEcukinumab) trial was designed to evaluate the efficacy of secukinumab in the management of axial manifestations of psoriatic arthritis (PsA).
Methods This phase 3b, double-blind, placebo-controlled, multi-centre 52-week trial included patients (≥18 years) diagnosed with PsA and classified by ClASsification criteria for Psoriatic Arthritis (CASPAR) criteria, with spinal pain Visual Analogue Score ≥40/100 and Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score ≥4 despite use of at least two non-steroidal anti-inflammatory drugs (NSAIDs). Patients were randomised (1:1:1) to secukinumab 300 mg, secukinumab 150 mg or placebo weekly for 4 weeks and every 4 weeks thereafter. At week 12, placebo patients were re-randomised to secukinumab 300/150 mg. Primary endpoint was ASAS20 (Assessment of SpondyloArthritis international Society) response with secukinumab 300 mg at week 12.
Results Patients were randomly assigned; 167 to secukinumab 300 mg, 165 to secukinumab 150 mg and 166 to placebo. Secukinumab 300 mg and 150 mg significantly improved ASAS20 response versus placebo at week 12 (63% and 66% vs 31% placebo). The OR (95% CI) comparing secukinumab 300 mg and 150 mg versus placebo, using a logistic regression model after multiple imputation, was 3.8 (2.4 and 6.1) and 4.4 (2.7 and 7.0; p<0.0001).
Conclusions Secukinumab 300 mg and 150 mg provided significant improvement in signs and symptoms of axial disease compared with placebo in patients with PsA and axial manifestations with inadequate response to NSAIDs.
Trial registration number NCT02721966.
- arthritis
- psoriatic
- antirheumatic agents
- biological therapy
- low back pain
- tumor necrosis factor inhibitors
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The data sets generated during and/or analysed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers’ access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved on the basis of scientific merit. All data provided is anonymised to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- arthritis
- psoriatic
- antirheumatic agents
- biological therapy
- low back pain
- tumor necrosis factor inhibitors
Key messages
What is already known about this subject?
Secukinumab, a fully human monoclonal antibody that directly inhibits interleukin (IL)-17A, has demonstrated significant and sustained efficacy across distinct clinical domains in active psoriatic arthritis (PsA). However, the efficacy of secukinumab or any other biological disease modifying anti-rheumatic drugs (bDMARDs) treatment specifically on axial manifestations in patients with PsA has never been investigated in a randomised controlled trial (RCT) setting.
What does this study add?
MAXIMISE (Managing AXIal Manifestations in psorIatic arthritis with SEcukinumab) is the first RCT to evaluate the efficacy of a bDMARD specifically in the management of the axial manifestations of PsA. Secukinumab 300 mg and 150 mg demonstrated significant improvements across the primary, key secondary and secondary clinical and imaging endpoints at week 12, which were sustained through week 52.
How might this impact on clinical practice or future developments?
The study provides evidence for the efficacy of IL-17A inhibition with secukinumab for the treatment of axial disease in patients with PsA. The results provide valuable data that will help inform treatment decision-making and deepen the clinical understanding of axial PsA, one of the disease manifestations lacking universally acceptable definition criteria.
Introduction
Spondyloarthritis (SpA) refers to a group of inter-related inflammatory musculoskeletal disorders that include either peripheral or axial SpA (axSpA). Psoriatic arthritis (PsA), the main type of peripheral involvement of SpA, is a heterogeneous, chronic, progressive, inflammatory condition, associated with enthesitis, dactylitis, skin and nail psoriasis that can affect peripheral joints but also the axial skeleton, with diverse patterns of involvement that can mimic different inflammatory arthritides.1 2 AxSpA is an inflammatory condition that can occur with (ankylosing spondylitis (AS) or radiographic axSpA) or without (non-radiographic axSpA) radiographic sacroiliitis. Although PsA and AS have a number of clinical features in common, AS accompanied with psoriasis and PsA with predominant axial involvement (axial PsA) are considered two separate disease entities with overlapping features.3 4 Axial PsA is not clearly defined, universally accepted criteria for axial PsA are currently lacking and the available outcome measures do not distinguish improvement of axial or peripheral symptoms.5 6 The development of classification criteria for axial PsA is currently being undertaken by a common effort of Assessment of SpondyloArthritis international Society (ASAS) and Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). The prevalence of axial disease in patients with PsA varies with disease duration and the definition used, occurring in 5% to 28% of patients with early-stage disease and in 25% to 70% of patients with long-standing PsA.3 Psoriatic nail dystrophy, the number of radiographically damaged joints, the number of swollen joints, the presence of periostitis and human leukocyte antigen (HLA)-B27 positivity have been identified as predictive factors associated with early or late axial involvement in previous reports.7–11 The burden of disease is underestimated in axial PsA because patients under-report axial symptoms as peripheral pain is more prominent and long-standing.11 12 In 2015, both the GRAPPA13 and the European League Against Rheumatism (EULAR) presented updated recommendations on the management of PsA.14 GRAPPA recommendations for the management of axial PsA are developed in accordance with the ASAS guidelines and suggest that biologics approved for axSpA may be used to inform treatment decisions for patients with axial PsA. The recently updated PsA EULAR recommendations are based on current practice and recommend therapy with a biological disease modifying anti-rheumatic drug (bDMARD), namely a tumour necrosis factor (TNF) inhibitor in patients with predominantly axial disease and an interleukin (IL)-17 inhibitor when there is relevant skin involvement.14 The recommendations from both groups note that the development of optimal recommendations for axial PsA remains a challenge.15
To the best of our knowledge, none of the randomised clinical trials performed to date that assessed the effect of biologics in PsA included a targeted assessment of axial disease. The only existing evidence comes from two observational studies based on clinical practice settings.10 11 Therefore, data from randomised controlled trials are lacking on the efficacy of biological treatment for the management of axial manifestations in patients with PsA.
Secukinumab, a fully human monoclonal antibody that directly inhibits IL-17A, has provided significant and sustained improvement in the signs and symptoms of active PsA and axSpA.16–18 The objective of the MAXIMISE (Managing AXIal Manifestations in psorIatic arthritis with SEcukinumab; NCT02721966) trial was to specifically evaluate the efficacy and safety of secukinumab 300 mg and 150 mg in managing axial manifestations in patients with PsA with an inadequate response to non-steroidal anti-inflammatory drugs (NSAIDs).
Methods
Study design
MAXIMISE was a phase 3b, double-blind, placebo-controlled, multi-centre 52-week trial that included 498 patients enrolled in 97 centres in Europe, Russia and Israel, between 3 October 2016 and 12 June 2018. The trial consisted of two treatment periods; a placebo-controlled period from baseline to week 12 followed by an active treatment period from week 12 to 52. After a screening period of up to 8 weeks, eligible patients were randomised (1:1:1) to subcutaneous (s.c.) secukinumab 300 mg, 150 mg or placebo weekly for 4 weeks and every 4 weeks thereafter. At week 12, placebo patients were re-randomised (1:1) to s.c. secukinumab 300 mg or 150 mg (online supplemental figure 1).
Supplemental material
Patients
Patients aged ≥18 years diagnosed with PsA and classified by ClASsification criteria for Psoriatic Arthritis (CASPAR) criteria, active spinal disease with a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score ≥4, spinal pain score ≥40 by Visual Analogue Score (VAS) (0 to 100 mm scale) and inadequate response to at least two NSAIDs over a 4-week period were included in the trial. Patients were excluded if they had a history of prior use of bDMARDs (such as TNF inhibitors, ustekinumab, IL-17, IL-23 inhibitors), active ongoing inflammatory conditions other than PsA, current treatment with conventional synthetic DMARDs other than methotrexate (MTX), and patients taking high potency opioid analgesics. Patients were allowed to continue prior use of NSAIDs, MTX and corticosteroids at enrolment through to the end of trial if on a stable dose from baseline to week 12.
Randomisation and masking
All eligible patients were randomised using Interactive Response Technology in a 1:1:1 ratio to secukinumab 300 mg, secukinumab 150 mg or placebo. At week 12, patients randomised to placebo at baseline were re-randomised in a 1:1 ratio to active treatment with secukinumab 300 mg or secukinumab 150 mg. Patients, investigators, site personnel and persons performing the assessments were blinded to the trial assignment (online supplemental material). To maintain blinding, all treatment groups received a consistent number of injections at each visit. The identity of the treatments were concealed by the use of study treatments in the form of pre-filled syringes for s.c. injection filled with secukinumab or placebo that were identical in appearance. Study treatments were administered by the patient or a caregiver after being instructed by site personnel.
Supplemental material
Outcome measures
Key efficacy, safety and tolerability assessments were done at screening, baseline, week 12 (primary endpoint), week 52 and time points in between. Protocol amendments are described in the online supplemental material. The primary endpoint was the proportion of patients achieving an ASAS20 response with secukinumab 300 mg at week 12. The ASAS Response Criteria (ASAS20) is defined as an improvement of ≥20% and ≥1 unit on a scale of 10 in at least three of the four main ASAS domains (namely patients global assessment (PtGA) of disease activity, PtGA of inflammatory back pain, BASFI (Bath Ankylosing Spondylitis Functional Index) and average of the last two questions on the six-question BASDAI) and no worsening of ≥20% and ≥1 unit on a scale of 10 in the remaining domain.19 The key secondary endpoint was an ASAS20 response with secukinumab 150 mg at week 12 after superiority of 300 mg was established. Other secondary endpoints were ASAS40, BASDAI50 and ACR20 (American College of Rheumatology) responses, mean change from baseline in spinal pain measured by VAS, Health Assessment Questionnaire Disability Index (HAQ-DI) score, Functional Assessment of Chronic Illness Therapy (FACIT)-fatigue scale and ASAS Health Index at week 12.
MRI of the spine and sacroiliac joints (SIJ) was performed at baseline and weeks 12 and 52 for all patients to assess sacroiliac and spinal inflammation as an exploratory endpoint to investigate whether these changes are affected by treatment with secukinumab. For patients who discontinued before or at week 12, an MRI was performed at the time of discontinuation. MRI scans were acquired using scanning techniques appropriate for the measurement of inflammation, bone marrow oedema and erosion20 and analysed centrally using the Berlin modification of the ASspiMRI scoring system (Berlin MRI score).20 MRI imaging of the spine and SIJ was implemented using a standardised scanning procedure monitored by a central imaging service agency to minimise differences among MRI scanners at different imaging centres. Spine images were acquired in two or three overlapping segments to achieve complete sagittal coverage of the spine (from C1 to S1). For SIJ, 3-plane localisers were acquired to have a true mid-sagittal slice showing the entire sacrum, based on which the centre of the joint space between S1 and S2 vertebral bodies was identified and 18 slices were prescribed in oblique coronal orientation. Details of the MRI image acquisition procedure are described in the online supplemental material.
The improvement in AS disease activity score (ASDAS) and Berlin MRI score for the spine and SIJ at week 12 to assess bone marrow oedema were exploratory outcome measures. ASAS20 response rates at week 12 were assessed in the subgroup of patients with positive MRI for spine and/or SIJ at baseline, as well as in the subgroup with or without concomitant MTX. Assessments at week 52 were ASAS20 and ASAS40, BASDAI50, spinal pain (VAS), ACR20, HAQ-DI, ASAS-Health Index, FACIT-fatigue and ASDAS. Safety analyses included all safety data reported up to and including the week 52 visit for each patient who received at least one dose of study drug.
Statistical analyses
Sample size was calculated based on a Fisher’s exact test assuming an overall type I error (two-sided) of 5%. To achieve 92% power and conservatively assuming a response rate of 40% in the placebo group, at least 150 patients per group were needed to be recruited under equal allocation to show a response rate of 60% in the secukinumab 300 mg group. Using the same number of patients per group, the second test had at least 80% power to detect a difference, if the true response rates are 57% in the secukinumab 150 mg group and 40% in placebo. To compensate for drop-outs and protocol violations, 165 patients per group (=495 in total) were required to be recruited into this trial. The full analysis set followed the intent-to-treat principle and comprised all patients from the randomised set to whom study treatment was assigned, fulfilling the clinical criteria for active axial disease, that is, spinal pain ≥40 and BASDAI ≥4. Patients were evaluated according to the treatment assigned at randomisation. The safety set included all patients who took at least one dose of study treatment during the entire treatment period. Summary statistics are presented for continuous demographic and baseline characteristic variables for each treatment group and for all patients in the randomised set, which included all patients originally randomised to secukinumab 300 mg or 150 mg and patients originally randomised to placebo who switched to secukinumab 300 mg or 150 mg at week 12 (placebo-secukinumab 300 mg or placebo-secukinumab 150 mg). Missing data up to week 12 for binary efficacy variables were handled using multiple imputation (MI) which imputes missing data based on patients’ actual data and observed data from similar patients in similar conditions. Analysis of covariance model was used to analyse continuous variables up to week 12. Data after week 12 through week 52 are reported as observed. Pre-defined exploratory analysis of the ASAS20/40 and BASDAI50 response at week 12 by Baseline Berlin MRI used the last observation carried forward (LOCF) method for imputation of missing data. LOCF technique was also undertaken as post-hoc analyses for ASAS20 and ASAS40 outcome measures.
Patient and public involvement
Patient or public were not involved in the design and conduct of the trial. The trial was conducted in accordance with the Declaration of Helsinki (General Assembly of the World Medical Association 2014) and was approved by institutional review boards or independent ethics committees at each participating centre. Written informed consent was obtained from all enrolled patients. Data were collected in accordance with Good Clinical Practice guidelines by the trial investigators and analysed by the sponsor.
Results
Patients
A total of 498 patients (167 to secukinumab 300 mg, 165 to secukinumab 150 mg and 166 to placebo group) were randomised; of these 425 (85%) patients completed the trial through week 52. The retention rates at week 52 were 83% (138/167) for secukinumab 300 mg, 86% (142/165) for secukinumab 150 mg, 89% (72/81) for placebo-secukinumab 300 mg and 91% (73/80) for placebo-secukinumab 150 mg; (figure 1). A total of 73 patients (15%) discontinued during the entire study period; with the most frequent reason being patient/guardian decision (33% (24/73)) followed by adverse events (AEs; 21% (15/73)) and lack of efficacy (15% (11/73)). Demographic and baseline disease characteristics and efficacy variables were comparable across groups (table 1). Patients had an established diagnosis of PsA with symptoms for around 7 years on average and around 50% were men. One or more of the parameters of inflammatory back pain were reported for the vast majority of the patients. Around 60% of patients had a positive MRI with inflammation in the spine and/or SIJ. HLA-B27 status, as reported by the investigator, was positive for 33% of the 261 patients for whom this data was available. Investigator reported X-ray data at baseline is summarised in online supplemental table S1. The mean time since last X-ray of SIJ ranged from 1.3 to 1.9 months across treatment arms and approximately two-third of the patient population had Grade 1 to Grade 4 sacroiliitis on either side.

Patient disposition. *5 patients were mis-randomised and were treated like screen failures for the data analysis. AE, adverse event; ICF, informed consent; Pt, patient; N, total number of randomised patients.
Demographics and baseline disease characteristics
Clinical efficacy
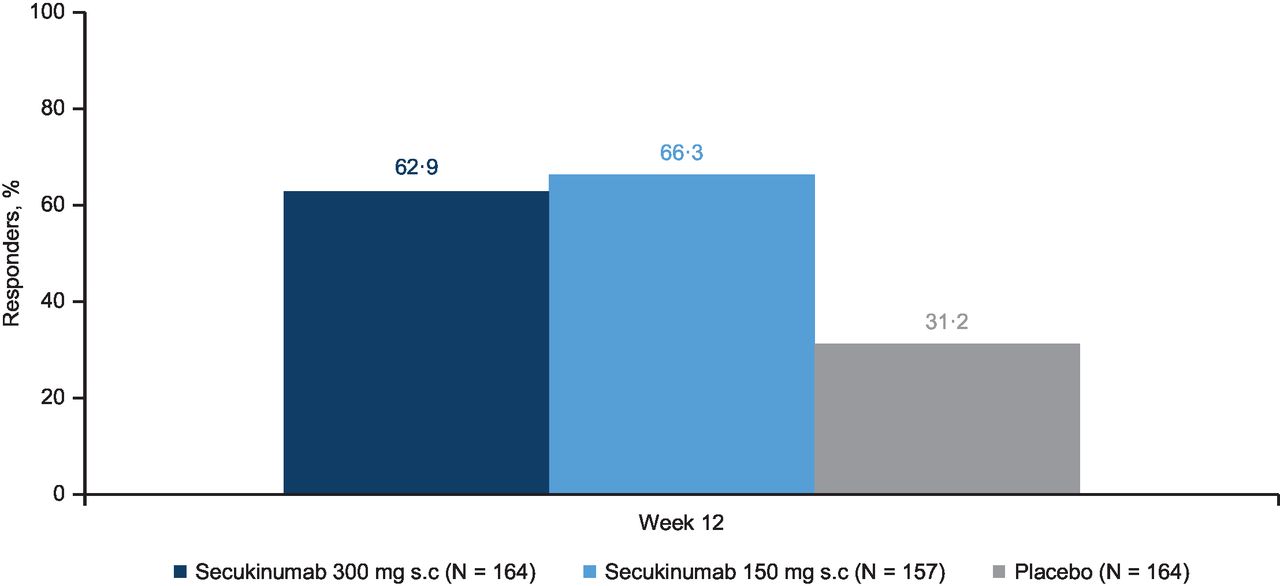
The primary and key secondary endpoints of the study were met; secukinumab 300 mg and 150 mg significantly improved ASAS20 response versus placebo at week 12 (63% and 66% vs 31% placebo). The OR (95% CI) for reaching ASAS20 response in the comparison of secukinumab 300 and 150 mg versus placebo, using a logistic regression model (with MI), was 3.8 (2.4 to 6.1) and 4.4 (2.7 to 7.0; p<0.0001; figure 2). ASAS40 response rates were greater with secukinumab 300 mg and 150 mg versus placebo at week 12 (44% (71/161) and 40% (60/151) vs 12% (20/161) placebo). The OR (95% CI) for reaching ASAS40 response in the comparison of secukinumab 300 mg and 150 mg versus placebo was 5.6 (3.2 to 9.8) and 4.7 (2.7 to 8.3), respectively (p<0.0001). Secukinumab improved other secondary endpoints at week 12 (table 2).

ASAS20 response rates at week 12 (MI). OR secukinumab 300 mg versus placebo: 3.8, p<0.0001 secukinumab 150 mg versus placebo: 4.4, p<0.0001. ASAS, Assessment of SpondyloArthritis International Society; MI, multiple imputation; N, total number of randomised patients (full analysis set); s.c., subcutaneous.
Other efficacy endpoints at weeks 12 and 52
The least square means (LSM) of treatment difference versus placebo in change from baseline in total Berlin MRI score for the entire spine at week 12 was −0.4 (0.1; secukinumab 300 mg; p<0.01) and −0.4 (0.1; secukinumab 150 mg; p<0.05). The LSM of treatment difference versus placebo in change from baseline in total Berlin MRI score for the SIJ at week 12 was −0.5 (0.2; secukinumab 300 mg; p<0.01) and −0.5 (0.2; secukinumab 150 mg; p<0.01). ASAS20 response rates at week 12 in the subgroup of patients with positive MRI for spine and/or SIJ at baseline were similar to the overall population, with 66% (58/88) for secukinumab 300 mg, 70% (51/73) for secukinumab 150 mg versus 27% (26/95) for placebo. ASAS20 response rates at week 12 in patients using concomitant MTX were 67% (secukinumab 300 mg), 67% (secukinumab 150 mg) versus 40% (placebo) and corresponding rates in the group without MTX use were 61%, 67% vs 25%. Pre-defined exploratory analysis of the ASAS20/40 and BASDAI50 response at week 12 by Baseline Berlin MRI score did not indicate a notable difference in the odds of achieving response between patients with either positive or negative Baseline Berlin MRIs in the secukinumab groups (online supplemental table S2).
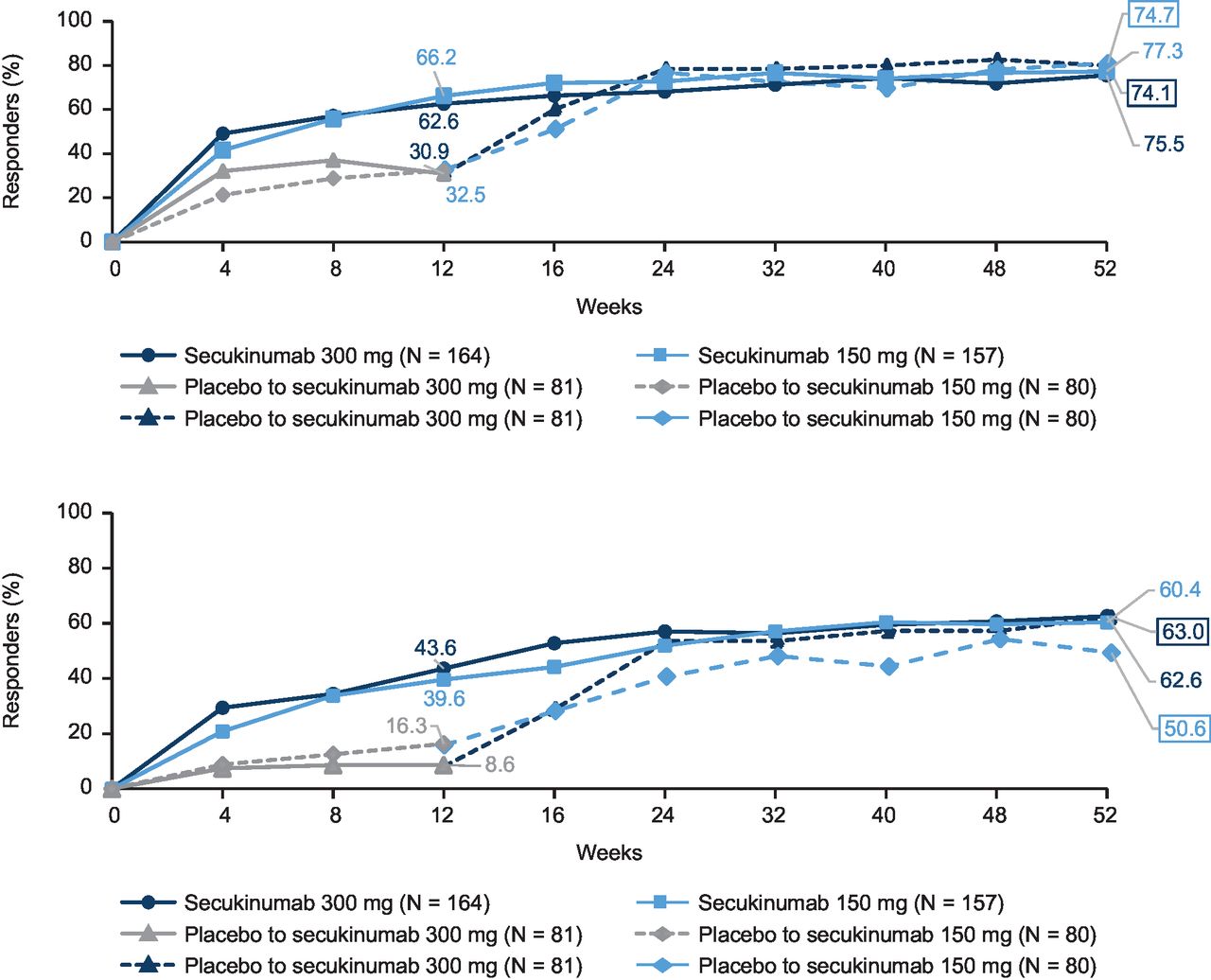
ASAS20 responses observed with secukinumab were sustained through week 52 and were 81% (113/139), 80% (113/141), 75% (54/72) and 80% (59/74) in the secukinumab 300 mg, 150 mg, placebo to secukinumab 300 mg and 150 mg groups, respectively. Reductions observed at week 12 in mean Berlin MRI score for the entire spine and SIJ were sustained at week 52. Notable reductions were also observed in placebo patients who switched to active treatment at week 12 (online supplemental figure S2). Other efficacy endpoints were sustained with secukinumab treatment through week 52 (table 2). ASAS20 response observed in the post-hoc analysis using LOCF was reported in 76% (123/163), 77% (119/154), 74% (60/81) and 75% (59/79) patients in the secukinumab 300 mg, 150 mg, placebo to secukinumab 300 mg and 150 mg groups, respectively. The corresponding rates for ASAS40 response using LOCF at week 52 respectively were 63% (102/163), 60% (93/154), 63% (51/81) and 51% (40/79) (figure 3). ASAS20 responses at week 52 in patients using concomitant MTX were 84% (secukinumab 300 mg), 82% (secukinumab 150 mg), 85% (placebo-secukinumab 300 mg) and 83% (placebo-secukinumab 150 mg); corresponding values in patients without concomitant MTX use were 80%, 79%, 66% and 77%, respectively.

{kind=link}
{kind=link}
{kind=link}
ASAS20 and ASAS40 responses through week 52*. (A) ASAS20. (B) ASAS40. *LOCF data in full analysis set. ASAS, Assessment of SpondyloArthritis international Society; LOCF, last observation carried forward.
Safety
The overall frequencies of non-serious AEs up to week 12 were reported in 65/167 (39%) and 60/165 (36%) patients in the secukinumab 300 mg and 150 mg groups, respectively, compared with 78/166 (47%) in the placebo group (table 3). The rate of serious AEs (SAEs) across secukinumab treatment groups over the entire treatment period was 28/493 (6%); none of the SAEs by preferred term were reported more than once in either of the secukinumab treatment groups (300 mg and 150 mg) over the entire treatment period. A total of seven serious infections (system organ class—infections and infestations) were reported over the entire treatment period. Three of these cases caused temporary dose interruption while the others did not warrant study treatment interruption. A total of eight cases of Candida infection (high level term) was reported. The cases of Candida infection were non-serious skin and mucosal infections of moderate severity and did not warrant study treatment interruption. One case of Crohn’s disease was reported through the entire treatment period with secukinumab (150 mg group), which led to study treatment discontinuation. Major adverse cardiovascular event was reported in three patients: one case each of ischaemic cardiomyopathy and cardiogenic shock, myocardial infarction (both in secukinumab 300 mg arm) and ischaemic stroke (secukinumab 150 mg arm). The event of ischaemic cardiomyopathy in a patient with a known history of hypercholesterolaemia and hypertension was fatal. Three cases of ‘malignant or unspecified tumour’ were reported through the entire treatment period. One was a case of small cell lung cancer (placebo-secukinumab 300 mg group), the second was a case of metastases to the spine (secukinumab 300 mg group) and the third was a case of adrenal neoplasm (secukinumab 150 mg) that was reported as benign by the investigator. One death (secukinumab 300 mg group) was reported in the trial which was a case of ischaemic cardiomyopathy in a 70-year-old male Caucasian patient with a known history of hypercholesterolaemia and hypertension that happened on day 204 and was not considered related to the study drug by the investigator (table 3).
Summary of secukinumab safety
Discussion
MAXIMISE is the first randomised controlled trial (RCT) to demonstrate the efficacy of a bDMARD in the management of the axial manifestations of PsA. Overall, significant improvements across multiple clinical and imaging endpoints were shown in a population with high activity of inflammatory back pain treated with secukinumab.
Treatment recommendations for axial PsA, extrapolated from AS and therapeutic interventions including new classes of biologics, have not reported efficacy in the axial manifestations of PsA in an RCT setting. However, in a previous study,9 in a combined cohort of patients with either PsA or AS from a single centre, 24% of the patients fulfilled the classification criteria (modified New York (mNY) or CASPAR) for both conditions indicating overlapping features of axial PsA and AS.
Nevertheless, the axial involvement, represents an unmet clinical need in determining the treatment strategy across all PsA manifestations and ultimately supports informed treatment decision-making. Additionally, patients with PsA tend to under-report axial symptoms and consequently the burden of disease might be underestimated for axial disease in such patients.11 12 As a consequence, the efficacy of a biological treatment in managing axial symptoms in PsA has been investigated only in two observational studies to date, and never in a randomised controlled setting.10–12
In the MAXIMISE trial, secukinumab 300 mg and 150 mg demonstrated significant improvements across all primary, key secondary and secondary endpoints at week 12. Clinical improvements were sustained through week 52 for the secukinumab arms; patients on placebo who switched to secukinumab 150 mg or 300 mg at week 12 improved rapidly and considerably across all assessed efficacy endpoints. In addition, MRI assessments demonstrated that secukinumab 300 mg and 150 mg significantly improved Berlin MRI scores versus placebo, providing objective evidence of reduced inflammation in both the spine and the SIJ for patients treated with secukinumab. Pre-defined exploratory analysis of the ASAS20/40 and BASDAI50 responses at week 12 by Baseline Berlin MRI score confirmed that MRI status at baseline did not have a significant effect on the outcome measures. The similar clinical responses in the MRI positive patients (for approximately 60% of the trial population) and the overall population regardless of MRI status at baseline further support the robustness of the clinical efficacy endpoints.
It is worth noting that the amount of active inflammation at baseline was lower compared with trials in active AS. However, the primary aim of the study was to assess the clinical outcomes of treatment with a bDMARD in axial PsA, and MRI positivity was not an inclusion criterion for the study. Nevertheless, since axial SpA and axial PsA may represent distinct disease entities although with overlapping features, such lower levels of objective signs of inflammation may be expected. Furthermore, the lack of a consensus in the definition of axial PsA has resulted in paucity of MRI data in axial PsA and hence there is no accurate benchmark of the expected levels of inflammation in terms of Berlin MRI score. It should also be noted that many studies have shown that MRI activity does not correlate with the burden of disease as measured by clinical assessments such as BASDAI, both for radiographic-axSpA and non-radiographic-axSpA) or, if at all, correlate very weakly with ASDAS before and after treatment.21 22
There is also an issue in PsA being a multifaceted condition as none of the available patient-reported outcomes (PROs) are specific to one domain, which is why MRI was assessed alongside the primary outcome of ASAS20 to allow an objective measurement of inflammation in the axial skeleton. However, it was decided not to mandate MRI changes to be included in the study to be as close as possible to the current clinical practice that is based on the clinical judgement of the treating physicians. Axial PsA is a poorly researched area, where further clinical insights, a universally accepted definition and disease specific endpoints are urgently needed. MAXIMISE, as the first randomised placebo-controlled study in this area, may provide clinically meaningful data on the treatment effects of a bDMARD on axial symptoms and a valuable data set to the research efforts on the classification and outcome measures of axial PsA.
The types and incidence of adverse events with secukinumab were comparable to placebo at week 12, with no apparent relation to dose. Over the entire treatment period, the rate of discontinuations due to adverse events and the rate of serious infections and Candidiasis was low for secukinumab and consistent with previously reported data for IL-17A inhibitors. One death occurred during the study. Overall, the safety profile of secukinumab was consistent with those published in previous reports.16–18
The limitations of the trial stem from the challenges in designing it; a major one being the lack of consensus in the clinical and/or imaging criteria to define this disease entity.4 5 In addition, axial PsA is distinct from axSpA3 and hence utilising mNY or ASAS criteria to determine the inclusion criteria for MAXIMISE would have been misinterpreted as having restricted its population to axSpA patients with psoriasis. Conversely, if stringent radiographic criteria had been applied, patients with clinical criteria of axial PsA without radiographic evidence would have been excluded and hence the results might have lacked generalisability to the whole axial PsA population.
Furthermore, the lack of axial PsA-specific outcome measures brought on the challenge of choosing the appropriate outcome measures. It is well-recognised that there is an unmet need for axial PsA specific outcome measures as ASAS and BASDAI although working well in AS trials, are not specific for axial inflammation in PsA. It should also be noted here that one of the ASAS response components is patient global assessment and BASDAI is impacted by the burden of peripheral arthritis, hence improvements in other domains of the disease may have influenced these results. The lack of randomised controlled trials and any precedent to aid the selection of axial PsA specific outcome measures, led to a general and inherent limitation. We therefore, selected ASAS20 as the primary endpoint as it was considered a valid option for a placebo-controlled randomised trial of this nature being the most frequently used outcome for assessing efficacy in axSpA trials. In addition, MAXIMISE included other assessments of axial symptoms such as ASAS40, BASDAI and ASDAS as secondary/exploratory outcomes and showed consistent results. Furthermore, greater improvements for both secukinumab 150 mg and 300 mg were shown versus placebo (p<0.0001) in the axial specific assessment of spinal pain indicating a clear effect of secukinumab on the axial skeleton. Finally, HLA-B27 data at baseline reported by the investigator was available for only 52% of the trial population.
In conclusion, secukinumab provided significant improvement in the signs and symptoms and objective signs of inflammation of axial disease in patients with psoriatic arthritis and inadequate response to NSAIDs. The clinical and imaging results from MAXIMISE provide valuable data that will support deepen the clinical understanding of axial PsA.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The data sets generated during and/or analysed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers’ access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved on the basis of scientific merit. All data provided is anonymised to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.
Acknowledgments
The authors thank the patients who participated in this study; the study investigators; John Gallagher, Novartis Pharmaceuticals UK Limited, London, UK, for valuable review. Dermot Whyms, Novartis Ireland Limited, Dublin, Ireland, has provided additional analytical and statistical review. Medical writing support, under the guidance of the authors, was provided by MK Vivek Sanker, Novartis Healthcare Private Limited, Hyderabad, India. The first draft of this manuscript was written by MK Vivek Sanker based on inputs from all the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors All authors were involved in the drafting and critical review of the manuscript and approved the final version for submission. LG, SJ and AM-V were involved in the acquisition of clinical data and participated as investigators in the clinical study. XB, LG, LCC, BS, MR and CP were involved in the conception or design of the study. EP, BS and KN were involved in the analysis of the data. All authors were involved with the interpretation of the results. All authors agreed to be accountable for all aspects of the work and attest to the accuracy and integrity of the work.
Funding The trial was designed by the sponsor, Novartis, in collaboration with the authors. The institutional review board at each participating centre approved the protocol. Data were collected in accordance with Good Clinical Practice guidelines by the study investigators and were analysed by the sponsor. All the authors contributed to the interpretation of the data and had access to the full data sets. Statistical analyses were performed by statisticians employed by the sponsor and were reviewed by all authors. Agreements between the sponsor and the investigators included provisions relating to confidentiality of the trial data. The writing support for the manuscript was provided by a medical writer from Novartis, India, and funded by the sponsor. All the authors vouch for the accuracy and completeness of the data and analyses, as well as for the fidelity of this report to the trial protocol, which are available from the funder.
Competing interests XB: Grant/research support from: AbbVie, and Novartis, Consultant for: AbbVie, BMS, Celgene, Chugai, Galapagos, Gilead, MSD, Novartis, Pfizer, and UCB; Speakers bureau: AbbVie, BMS, Celgene, Chugai, MSD, Novartis, Pfizer, and UCB. LG: Research grants: Amgen, Lilly, Janssen, Pfizer, Sandoz, Sanofi, and Galapagos; consulting fees: AbbVie, Amgen, BMS, Biogen, Celgene, Gilead, Janssen, Lilly, Novartis, Pfizer, Samsung Bioepis, Sanofi-Aventis and UCB. EP: Employee of Novartis with Novartis stock. SJ: Research support/speaker: AbbVie, Pfizer, Roche, Novartis, MSD, Sandoz, Lilly, Egis, UCB and Celgene. AM-V: None declared. SD: Consultant for AbbVie, Biogen, BMS, Celgene, Lilly, MSD, Novartis and UCB; Speakers bureau: AbbVie, BMS and Celgene, Lilly, Novartis, Pfizer and Sanofi. BS: Employee of Novartis. MR: Employee of Novartis with Novartis stock. KN: Employee of Novartis. CP: Employee of Novartis with Novartis stock. LCC: Grant/research support: AbbVie, Janssen, Lilly, Novartis and Pfizer; Consultant for: AbbVie, Amgen, Biogen, Celgene, Pfizer, UCB, Boehringer Ingelheim, Novartis, Lilly, Janssen, Sun Pharma, Prothena and Gilead.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.