Article Text

Abstract
Background Recently, we documented the likely non-inferiority of Combinatietherapie Bij Reumatoïde Artritis (COBRA)-light therapy (methotrexate increased to 25 mg/week with initial prednisolone 30 mg/day) compared with the original COBRA therapy (methotrexate 7.5 mg/week, sulfasalazine 2 g/day, with initial prednisolone 60 mg/day) after 26 weeks in patients with early active rheumatoid arthritis (RA).
Objective To assess the non-inferiority of COBRA-light versus COBRA after 1 year in terms of disease activity (DAS44), functional outcome (Health Assessment Questionnaire (HAQ)) and radiographic progression (Sharp/van der Heijde score (SHS)), and to assess the effect of adding etanercept.
Methods An open-label, randomised controlled, non-inferiority trial of 162 patients with active early RA, following a treat-to-target protocol incorporating the addition of etanercept if DAS44 ≥1.6 at weeks 26 or 39.
Results Both groups showed major improvements in DAS44 after 52 weeks: mean (SD) −2.41 (1.2) in the COBRA and −2.02 (1.0) in the COBRA-light group (p=ns). In both groups, functional ability improved and radiological progression of joints was minimal. At least one adverse event was reported in 96% of the patients in both groups. In total, 25 serious adverse events occurred: 9 vs 16 in COBRA and COBRA-light, respectively. Treatment actually instituted was often less intensive than required by the protocol: of the total population, 108 patients (67%) required etanercept (more in the COBRA-light group), but only 67 of these (62%) actually received it.
Conclusions Intensive COBRA or COBRA-light therapy has major, comparably favourable effects on disease activity, functional ability and radiological outcome after 1 year in patients with early RA. Protocolised addition of etanercept was often not implemented by treating rheumatologists, and patients receiving it appeared to have limited added benefit, probably because of low disease activity levels at its initiation.
Trial registration number: ISRCTN55552928.
- Early Rheumatoid Arthritis
- Anti-TNF
- DMARDs (synthetic)
- Outcomes research
- Treatment
Statistics from Altmetric.com
Introduction
Treatment with disease-modifying anti-rheumatic drugs (DMARDs) has proven to be effective in the treatment of rheumatoid arthritis (RA) in improving disease activity and physical functioning and preventing joint progression.1–7 Patients with early RA are usually treated with methotrexate (MTX), often combined with glucocorticoids, with sulfasalazine and/or hydroxychloroquine as alternatives.8 A very effective strategy is COBRA therapy (COmbinatietherapie Bij Reumatoïde Artritis (COBRA)) in which patients receive MTX (7.5 mg/week), sulfasalazine (2 g/day) and initially high-dose prednisolone (60 mg/day). In the BeSt trial, this strategy proved to be as effective and safe as the combination of high-dose MTX (25 mg/week) and anti-tumour necrosis factor (TNF) treatment (infliximab).9–11 Despite this, rheumatologists have reservations in prescribing COBRA therapy for several reasons, including concerns over the possible side effects of high-dose or prolonged glucocorticoid exposure.12 ,13 Therefore, we recently introduced COBRA-light therapy, combining MTX (25 mg/week) with an initially lower dose of prednisolone (30 mg/day). Our first report of the COBRA-light trial, in which both therapies were applied in treat-to-target strategies for patients with early RA, suggested that COBRA-light was non-inferior to COBRA in clinical outcomes, safety and efficacy after 26 weeks of treatment.14
The COBRA-light trial protocol specified 1 year of treatment, in which patients were to start etanercept if they did not reach minimal disease activity at week 26 or 39. This report describes clinical and radiological outcomes at 1 year.
Patients and methods
From March 2008 to April 2011, 164 DMARD-naïve Dutch patients with recent-onset RA were included in the COBRA-light trial. This study was a multicentre, randomised, open-label trial (http://www.controlled-trials.com; ISRCTN55552928). Patients were randomised to the COBRA-light or COBRA strategy (as applied in the BeSt trial), to determine the non-inferiority of COBRA-light therapy. The treatment goal was minimal disease activity (at that time defined as clinical remission: Disease Activity Score in 44 joints (DAS44) <1.6), and treatment intensification of MTX and addition of etanercept to reach this goal was protocolised up to 52 weeks (see online supplementary file appendix 1). Patient-selection criteria, the randomisation process and study design have been reported previously.14
Medical ethics committees at each participating centre approved the protocol; patients gave written informed consent before inclusion, and the study was conducted in accordance with the Declaration of Helsinki/Good Clinical Practice.
Clinical assessments
Outcome measures were disease activity variables, factors to calculate different response and remission criteria, and several patient-reported outcomes, including the changed and achieved state in physical functioning as measured by the Dutch version of the Health Assessment Questionnaire (NL-HAQ).15–18 Medication use was ascertained by self-report of the patients through patient diaries and from patient medical records.14
Intra-articular and intramuscular glucocorticoid injections were allowed. To correct for possible influence of these injections on disease activity assessment, the score of joints injected in the 12 weeks before a study visit was set to swollen and painful (Ritchie articular index score 1), if no score was recorded. The DAS44 score of patients receiving an intramuscular injection in the 4 weeks before a study visit was set to missing.
All adverse events and protocol violations were reported at each visit by active solicitation.14 An independent committee of two rheumatologists who were not involved in the execution of the study evaluated all adverse events to identify serious adverse events (SAEs) according to the standard of the Directive 2001/20/EC of 4 April 2001,19 and assessed their potential relationship to treatment. This same committee classified all protocol violations as major or minor and confirmed protocolised treatment deviations. A major protocol violation was defined as any modification from protocol, design or procedure that could affect the completeness, accuracy, reliability or integrity of the study data. These violations were further classified as lack of compliance of the physician or the patient, administrative mistakes, and other reasons. Minor violations were defined as small or temporary alterations in treatment deemed not to affect the study data. Protocolised treatment deviations were changes in protocol treatment (dose reduction or cessation) due to adverse events or secondary ineligibility (eg, the desire to become pregnant).
Radiology
Radiographs were obtained at baseline, week 26 and at 1 year of follow-up, and scored by two independent trained assessors according to the Sharp/van der Heijde score (SHS).20 The assessors were blinded to group allocation and patient identification, but were aware of time order, as reading in chronological order was carried out; this is most sensitive for detecting progression and leads to less bias.21 If assessors differed by more than 5 points in the SHS, a third assessor (DvS) was consulted to reach a consensus. The intraclass correlation coefficient agreement between the two assessors was 0.90 (95% CI 0.8 to 1.0), assessed on a test sample of 30 patients. The mean of the two observers’ scores was reported. If radiographs were missing at week 26, and scores at baseline and week 52 remained stable, the same score was imputed at week 26. For missing radiographs at week 52, SHS was extrapolated with linear imputation, using scores at baseline and week 26. Missing baseline scores (n=1) were imputed by retrograde extrapolation from scores at week 26 and 52.
Anti-TNF therapy
The protocol required treatment intensification with etanercept for patients who did not reach a DAS44 <1.6 at week 26 or 39. In the COBRA group, MTX dose was initially increased to 25 mg/week in 2-weekly steps, followed by etanercept 50 mg once weekly subcutaneously if minimal disease activity had not been achieved at the next assessment (see online supplementary file appendix 1). In the COBRA-light group, the initial step was the possibility of switching to subcutaneous MTX followed by etanercept. Patients received etanercept until week 52 (period of 3–6 months depending on start date). During etanercept use, DMARD dosages were kept stable, except in the case of adverse events and in cases of minimal disease activity, when prednisolone was tapered to 0 mg/day.
Statistical analysis
Non-inferiority was defined as a difference in ΔDAS44 between the two groups of 0.5 points or less. All patients who received at least one medication dose were included in the modified intention-to-treat (ITT) protocol. Data are presented as mean±SD, or as median (IQR) in the case of skewed distribution.
All disease activity outcome measures with Gaussian distribution were analysed with Generalised Estimating Equation (GEE) to assess differences between treatment strategies over time and to correct for repeated measures. Baseline measures were included in all analyses to correct for regression to the mean. Change in DAS44 over time was analysed as outcome, treatment group and baseline DAS44 as explanatory measures. Measures with a skewed contribution were first log-transformed (natural logarithm) and then analysed with GEE. A linear regression model was used to assess difference in radiological progression and for results expressed as area under the curve. Differences in proportions of patients achieving remission and starting etanercept were expressed as relative risks (RRs). χ2 for trend analyses were performed on American College of Rheumatology (ACR) response (ACR20 but not ACR50, ACR50 but not ACR70, and ACR70) and European League against Rheumatism (EULAR) response (non-responder, moderate responder but not good responder, good responder).22 All statistical analyses were conducted using IBM SPSS statistics V.20. A two-sided p<0.05 was considered significant. For the primary outcome (change in DAS44), non-inferiority of COBRA-light can be declared if the upper limit of the 95% CI (two-sided) around the difference between the groups is <0.5.23 ,24 For all other outcomes, acceptance of the null hypothesis was deemed to document non-inferiority.
Results
A total of 164 patients were included: 81 receiving COBRA therapy and 83 COBRA-light therapy (figure 1). Two patients did not start treatment because of withdrawal of informed consent immediately after randomisation. These patients were not included in the ITT analyses. Baseline demographic and clinical characteristics for the two groups were similar, except for a small difference in baseline DAS44: mean (SD) 4.09 (0.7) for COBRA and 3.96 (0.9) for COBRA-light, as shown in table 1.14 Half of all patients were positive for both rheumatoid factor and anti-cyclic citrullinated peptide.
Demographic and disease activity measures at baseline and at 52 weeks

Flow chart of Combinatietherapie Bij Reumatoïde Artritis (COBRA)-light therapy trial. In the COBRA group, one patient discontinued at week 13 (myocardial infarction), one at week 26 (intolerance of medication), and one at week 39 (bilateral pulmonary embolism). In the COBRA-light group, one patient discontinued at week 13 (poor compliance with treatment) and three patients at week 26 (manic episode, desire to become pregnant, poor compliance with treatment). All patients were alive at week 52 of the trial. The nine patients who did not start or prematurely stopped the trial were older (mean (SD) 59 (12.3) years), had a shorter disease duration (median 13 weeks, IQR 6–18.5), a lower Disease Activity Score in 44 joints (3.94 (0.4)) and a higher Health Assessment Questionnaire score (median 1.88, IQR 1.32–2.13) at baseline compared with the patients with complete follow-up.
Seven patients dropped out during the trial: three in the COBRA group and four in the COBRA-light group (figure 1). Ten patients (COBRA, 3; COBRA-light, 7) received an intra-articular injection, five in the 12 weeks before an assessment. Nine patients received an intramuscular glucocorticoid injection (COBRA, 4; COBRA-light, 5), one within 4 weeks of an assessment.
Clinical outcomes
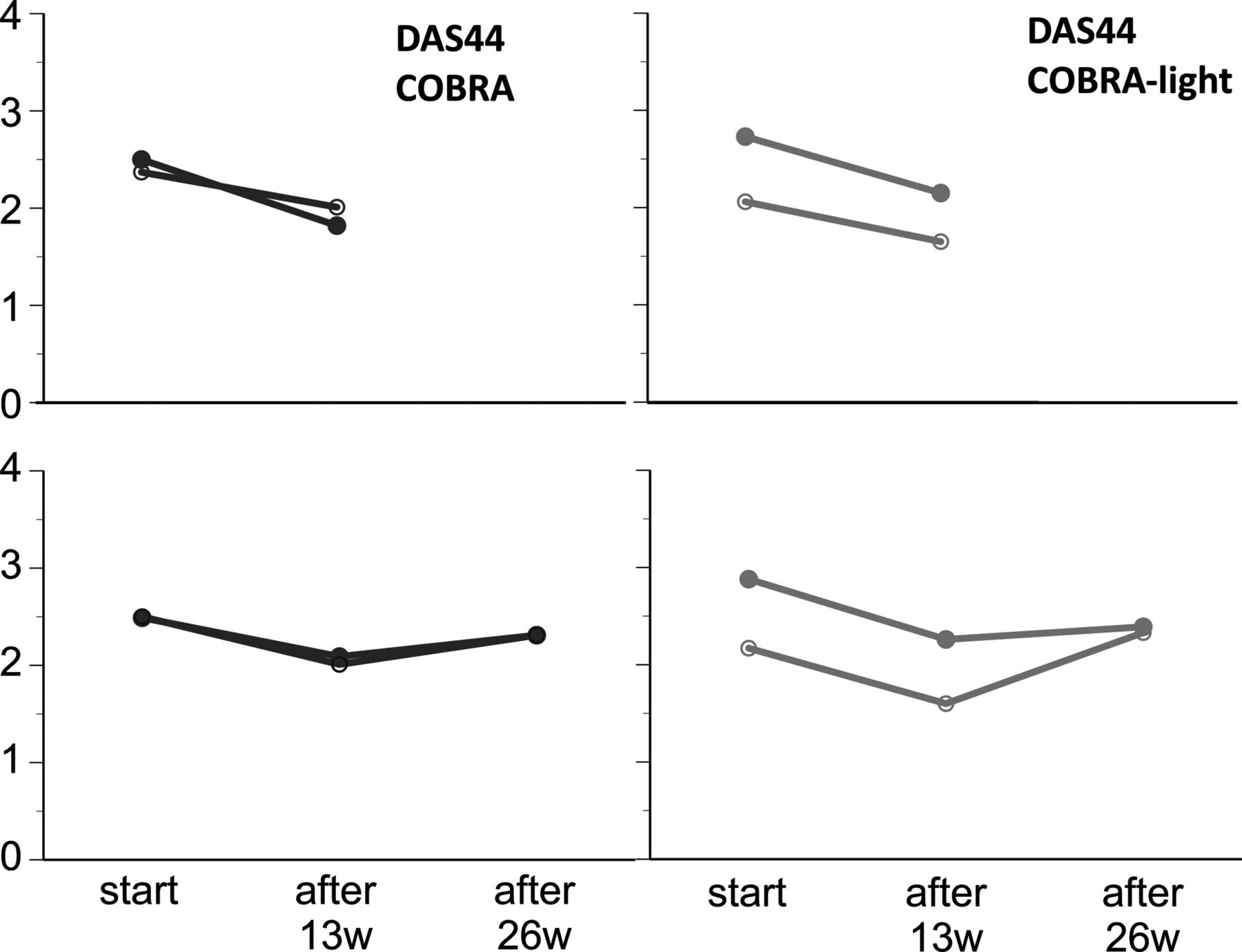
Both groups showed major improvements in disease activity between baseline and week 26 as reported previously,14 with stabilisation in the second 6 months; COBRA-light remained non-inferior to COBRA. Longitudinal change over time in DAS44 between baseline and week 52 was −2.41 (1.2) in the COBRA group and −2.02 (1.0) in the COBRA-light group (adjusted β=0.19 (0.1), 95% CI −0.07 to 0.45, p=0.15; table 1, figure 2). At this time point, the CI of difference in ΔDAS44 does not include the predefined clinically relevant threshold of 0.5. Minimal disease activity (DAS44 <1.6) was reached in 38% of the patients in COBRA and 31% in COBRA-light (ACR/EULAR remission in 15% vs 17%, respectively) (table 1).

Mean change in outcomes of treatment. Data are expressed as mean (SD). Black line, COBRA therapy; grey line, COBRA-light therapy. N=74 or greater at all time points. COBRA, COmbinatietherapie Bij Reumatoïde Artritis; CRP, C-reactive protein; DAS44, Disease Activity Score for 44 joints; ESR, erythrocyte sedimentation rate; HAQ, Health Asessment Questionnaire.
The two strategies had an almost equally positive effect on functional ability: decrease of 0.79 and 0.76 (0.7) points on the HAQ for COBRA and COBRA-light, respectively, and showed consistent patterns in single disease activity measures over time, except for a larger erythrocyte sedimentation rate (ESR) decrease for COBRA (table 1). At week 26 and 39, ∼40% of the patients in both groups could taper prednisolone to 0 mg/day.
No significant differences were found in any of the other outcomes, supporting non-inferiority. Differences in proportions of patients reaching EULAR response, ACR20, ACR50 and ACR70 improvement were small and did not favour one treatment group (table 1). At week 52, 6% of the patients were EULAR non-responders in both groups. This proportion increased when the cut-off point for non-responders was set as HAQ score ≥1.0 at week 52: 17% in COBRA and 30% in COBRA-light. Non-responders (DAS44 ≥2.4 or HAQ score ≥1.0 or ΔSHS ≥5) had a somewhat higher baseline DAS44 than responders (∼4.2 vs 3.9, respectively). This was mostly driven by a high visual analogue scale (more than 65 points) and approximately 23 tender joints (data not shown).
Radiology
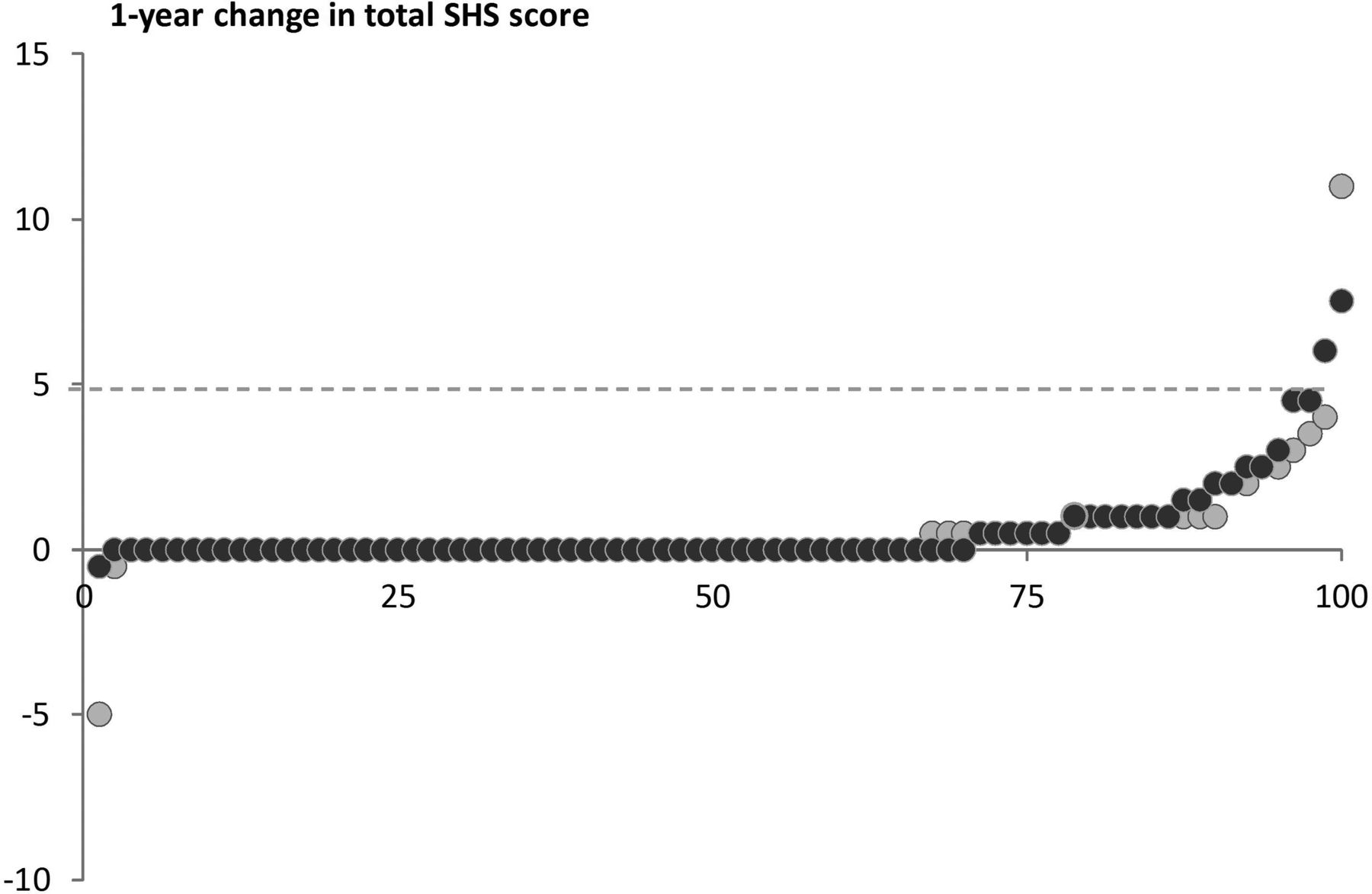
In 18 patient sets, the two assessors differed by at least 5 points, and the score of the third assessor was applied. The intraclass correlation coefficient agreement, based on the mean total SHS at baseline between the two assessors in this dataset (excluding patients with a consensus score), was 0.91 (95% CI 0.88 to 0.93). At baseline, the SHS was 2.66 (6.5) for COBRA and 1.61 (4.0) for COBRA-light (table 1); erosive disease, as defined by the 2010 ACR/EULAR criteria,25 was present in 7% vs 2% in COBRA and COBRA-light, respectively. At week 52, the increase in SHS was 0.49 (1.6) and 0.59 (1.4) points for COBRA and COBRA-light, respectively (p=0.42), with no difference in the score components. In COBRA, 6% had erosive disease, compared with 7% in COBRA-light. An increase in damage of 5 points or more occurred in only 5% of the patients (figure 3).

Probability plot Sharp/van der Heijde score (SHS). Black circles, COBRA therapy; grey circles, COBRA-light therapy. COBRA, COmbinatietherapie Bij Reumatoïde Artritis.
Anti-TNF therapy
Per protocol (DAS44 ≥1.6) at either week 26 or 39, fewer COBRA than COBRA-light patients needed treatment intensification with etanercept: 47 (59%) vs 61 (75%) (RR 1.67, 95% CI 1.05 to 2.65, p=0.03). If the protocol cut-off had been set at DAS44 ≥2.4 (‘low disease activity’, corresponding to DAS28 ≥3.2), 38% versus 49%, respectively, would have needed intensification to etanercept (p=0.04). However, owing to protocolised treatment deviations and/or protocol violations, only 27 of 47 (57%) COBRA patients versus 40 of 61 (66%) COBRA-light patients actually started etanercept (p=0.04). In the COBRA group, 16 and 11 patients started at week 26 and 39, respectively; one patient started at week 30 because of a rapid increase in disease activity between week 26 and 39. In the COBRA-light group, 28 and 10 patients started at week 26 and 39, respectively. In addition, two patients started at week 13 because of high disease activity (DAS44=6.0 and 4.5). The two groups had the same minimal progression of ΔSHS as the total group and were randomly distributed on the probability plot in figure 3 (data not shown). Etanercept was, in general, well tolerated: at week 39, adverse events had occurred in 24% of etanercept users compared with 13% of non-users. At week 52, proportions were 26% vs 14%, respectively. The 22 COBRA patients and 21 COBRA-light patients who were etanercept non-starters had somewhat lower disease activity than actual starters, as evidenced by DAS44. These differences were only statistically significant in the COBRA-light group (table 2). In addition, in 60% of these cases, the physician assessed the disease activity to be lower than did the research nurse (data based on remarks of physicians during collection of protocol violations).
Etanercept data
Thirteen weeks after the indication to start etanercept, the mean disease activity had decreased in all groups regardless of whether etanercept was actually started: in the COBRA group, DAS44 had decreased by 0.57 (0.8) and 0.23 (0.5) points for actual starters and non-starters, respectively (adjusted β=0.29 (0.24), 95% CI −0.21 to 0.78, p=0.25); in the COBRA-light group, it had decreased by 0.55 (0.8) and 0.29 (0.9) points, respectively (adjusted β=−0.05 (0.23), 95% CI −0.51 to 0.41, p=0.83) (figure 4). Of the actual starters, 27% could taper prednisolone to 0 compared with 35% of the non-starters (p=ns).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean change in Disease Activity Score in 44 joints (DAS44) for treatment in etanercept-intended vs actual starters. Data are expressed as mean (SD). Black line, COBRA therapy; grey line, COBRA-light therapy. Etanercept starters (closed circles): n=27 and 40 for COBRA and COBRA-light respectively. Etanercept nonstarters (open circles): n=13 and 10 respectively. COBRA, COmbinatietherapie Bij Reumatoïde Artritis.
A total of 46 patients received etanercept for 26 weeks (16 in the COBRA group and 30 in the COBRA-light group); in this group, DAS44 had increased again between week 39 and 52, resulting in a net decrease of 0.31 points in COBRA and 0.36 points in COBRA-light therapy over the total 6-month period.
Protocol violations and adverse events
After 52 weeks of treatment, nearly all patients reported at least one adverse event after active solicitation: 96% in both groups. The most commonly reported were skin problems, mild gastrointestinal problems, and infections; their incidence did not differ between the two groups (see online supplementary file appendix 2). Patients in both groups gained ∼1.0 kg in weight (p=0.89). In total, 16 patients (20%) in the COBRA arm had an increase of >5 kg in weight compared with 11 patients (14%) in the COBRA-light arm (p=0.31). Only one patient was newly diagnosed with diabetes mellitus type II and needed treatment with oral antidiabetic drugs (COBRA group). Nine patients needed treatment for hypertension (five in the COBRA group and four in the COBRA-light group), and eight patients needed treatment for dyslipidaemia (three and five in the COBRA and COBRA-light groups, respectively).
In total, 25 SAEs occurred: nine and sixteen in COBRA and COBRA-light patients, respectively. The COBRA patients experienced the following SAEs: myocardial infarction, pulmonary embolism, hospitalisation for pneumonia, planned cataract surgery on both eyes (2×), planned surgery of the cervical spine, attempted suicide due to depression, fibula fracture caused by MTX osteopathy, and a pelvis fracture. SAEs in the COBRA-light patients included lung carcinoma (2×), planned knee replacement surgery (2×), planned hallux valgus surgery, planned varicose vein surgery, planned control coloscopy for diverticulitis, hospitalisation for arrhythmia, manic episode, replacement and hospitalisation for a retina bleeding, planned surgery for cyst removal, hospitalisation for anaemia caused by duodenal ulcers, planned cholecystectomy, surgery for an inguinal hernia, hospitalisation and surgery for a hip fracture after a fall, and surgery for chronic synovitis.
Over the year, there were one or more protocol violations/deviations for more than half of the patients: 49 in COBRA and 47 in COBRA-light. More major violations occurred in the COBRA than the COBRA-light group: 68 vs 48 (see online supplementary file appendix 2). Most of these violations were due to non-compliance of the physician with the protocol, and most of the violations led to a decrease in intensity of the therapy.
Discussion
This study confirms that, in patients with early RA, COBRA and COBRA-light therapy are similarly efficacious in suppressing clinical disease activity and improving functional ability. In addition, both strategies have been shown to effectively suppress progression of joint damage after 52 weeks of treatment. The addition of the TNF-blocking agent, etanercept, to the treatment had limited added benefit in decreasing disease activity.
The present effects are comparable to other treat-to-target studies in patients with early RA.3 ,6 ,10 ,26 In the BeSt trial, the goal of low disease activity (DAS44 ≤2.4) was reached in 71% of the patients receiving COBRA therapy; in the present study, 73% reached this goal compared with 67% receiving COBRA-light therapy. Mean rates of damage progression were only slightly above zero, and this must be interpreted in light of the method of reading in a known sequence. This method results in slightly higher rates than with random reading, the method used in most trials of biological agents.27
The treatment goal in this trial was set at DAS44 <1.6; with a combination of traditional DMARDs, 47% and 38% of patients for COBRA and COBRA-light therapy, respectively, achieved this, compared with 32% in the BeSt trial. Remarkably, although 67% of our study population required etanercept treatment according to the protocol, only 62% of those patients actually received it. It is possible that our predefined goal was too stringent compared with daily practice, where lack of low disease activity or severe disease activity (threshold DAS28 ≥3.2, corresponding to DAS44 ≥2.4) is used as an indication to start biological agents. We also noticed that treating physicians sometimes did not agree with the DAS44 assessors. Therefore, treatment was not intensified, either because physicians assessed patients with less active disease or both patient and physician were willing to accept low disease activity balanced against the risk and costs of biological agents—for instance, TNF-blocking agents. This was also observed earlier in the DREAM study.28
Our data show that a low DAS44 threshold in treat-to-target protocols poses challenges in clinical practice, because the measure is not reliable in patients with low disease activity, mostly because of its sensitivity to small changes in ESR and global health scores, when joint scores are low or zero.29 One option may be to add a biomarker in the assessment of disease activity. For example, in a pilot trial that added a biomarker in the assessment of disease activity (measured with DAS28), 90% of the patients had reached minimal disease activity (DAS28 <2.6) after 40 weeks of treatment with a modified COBRA strategy followed by infliximab intensification, according to CTX2 values.30
Nevertheless, the many protocol deviations and violations preclude a good assessment of the effect of etanercept in the proposed treatment strategy. Of interest, the added benefit of etanercept in actual starters appeared to be rather limited: both actual starters and non-starters ended up with a mean DAS44 score of 2.2 at week 52. To our knowledge, this study is the first to assess the effect of adding anti-TNF therapy after 6 or 9 months of treatment with intensive combination regimens including prednisolone; a study in a real world setting also found lack of good outcomes explained by biological agents.31 Other trials starting with an initial treatment with biological agents and DMARDs had comparable results.6 ,10 In the NEO-RACO trial, patients treated with traditional DMARDS and an initial dose of infliximab showed equivalent results on disease measures after 6 months, compared with patients treated with traditional DMARDs and placebo.6 In the BeSt trial, the infliximab plus MTX treatment arm showed similar results to the COBRA arm.10 In the SWEFOT trial, infliximab treatment after insufficient reaction with 3–4 months of MTX use resulted in a significant decrease in disease activity levels, as well as in the TEMPO trial with etanercept.32 ,33 Patients in these trials had much higher disease activity levels at the start of treatment with a biological agent compared with our patients. These data illustrate the well-known phenomenon that anti-TNF therapy is effective for treating patients with high disease activity levels, but might have limited benefit in patients with low disease activity, as suggested in our trial.
This study has limitations. The first is the open-label design: trained research nurses, who are not involved in routine care, assessed all outcomes to minimise any influence on the outcome measures. Of course, in classical trial designs, blinding would have limited bias of the results. On the other hand, this design is closer to daily practice, which increases its external validity. Second, power calculations were primarily based on week 26 outcome; therefore, the outcome after 52 weeks is a secondary outcome, but is, nevertheless, clinically very relevant. However, the clinically relevant difference in ΔDAS44 was still <0.5 at week 52, as for the primary outcome time point at week 26. Therefore, we think our non-inferiority claim still holds. Several other limitations that have previously been mentioned14 (eg, non-inferiority, protocol violations) also apply to this study.
Although most patients responded with both strategies, 6% were still EULAR non-responders at week 52. Corresponding proportions of HAQ non-responders (ie, HAQ score ≥1.0) at week 52 were noted: 17% and 30% for COBRA and COBRA-light therapy, respectively. In future, these patients should be identified earlier, as they probably require different treatment from that usually provided.
In conclusion, addition initially of moderate-dose prednisolone to intensive MTX treatment as proposed in the COBRA-light therapy yields similar results to the original COBRA therapy, with strongly positive effects on disease activity, functional ability and progression of joint damage in early RA. Further intensification with etanercept after an inadequate response, but mostly in patients with low disease activity, was often unacceptable to patients and physicians, leading to non-compliance with the protocol. In patients who actually started etanercept, its added value appeared to be limited, probably because of low disease activity at its initiation.
Acknowledgments
We would like to thank all patients, as well as all doctors who enrolled patients in this study, and all research nurses who were involved in patient management. We also thank B Blomjous and E Kooijmans, for their work over the past few years entering data into the COBRA-light therapy database, Dr L Burgemeister and Dr M Gerritsen, for their work as an independent committee judging protocol deviations and SAEs, and Dr K Britsemmer and Dr J van Nies, for scoring all radiographs.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix 1
- Data supplement 2 - Online appendix 2
Footnotes
Handling editor Tore K Kvien
Contributors All authors substantially contributed to the conception or design of the study and the acquisition, analysis and interpretation of data. All revised the article critically and approved the final version to be published. All agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This research was performed within the framework of project T1-106 of the Dutch Top Institute group, and additionally funded by an unrestricted grant from Pfizer.
Competing interests WFL has received speaker’s fees from Merck, Abbvie, Roche and Pfizer. MN has received research grants from Abbvie, BMS, MSD, Pfizer, UCB and Roche. He has also acted as a consultant for Abbvie, BMS, Pfizer and Roche. Furthermore, he has participated in speaker bureaus for Abbvie, BMS, Pfizer and Roche.
Ethics approval Medical ethics committees at each participating centre approved the protocol; patients gave written informed consent before inclusion, and the study was conducted in accordance with the Declaration of Helsinki/Good Clinical Practice.
Provenance and peer review Not commissioned; externally peer reviewed.