Article Text

Abstract
Objective To identify genetic associations with severity of radiographic damage in ankylosing spondylitis (AS).
Method We studied 1537 AS cases of European descent; all fulfilled the modified New York Criteria. Radiographic severity was assessed from digitised lateral radiographs of the cervical and lumbar spine using the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS). A two-phase genotyping design was used. In phase 1, 498 single nucleotide polymorphisms (SNPs) were genotyped in 688 cases; these were selected to capture >90% of the common haplotypic variation in the exons, exon–intron boundaries, and 5 kb flanking DNA in the 5′ and 3′ UTR of 74 genes involved in anabolic or catabolic bone pathways. In phase 2, 15 SNPs exhibiting p<0.05 were genotyped in a further cohort of 830 AS cases; results were analysed both separately and in combination with the discovery phase data. Association was tested by contingency tables after separating the samples into ‘mild’ and ‘severe’ groups, defined as the bottom and top 40% by mSASSS, adjusted for gender and disease duration.
Results Experiment-wise association was observed with the SNP rs8092336 (combined OR 0.32, p=1.2×10−5), which lies within RANK (receptor activator of NFκB), a gene involved in osteoclastogenesis, and in the interaction between T cells and dendritic cells. Association was also found with the SNP rs1236913 in PTGS1 (prostaglandin-endoperoxide synthase 1, cyclooxygenase 1), giving an OR of 0.53 (p=2.6×10−3). There was no observed association between radiographic severity and HLA-B*27.
Conclusions These findings support roles for bone resorption and prostaglandins pathways in the osteoproliferative changes in AS.
- Ankylosing spondylitis
- genetic association study
- x-ray
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS) is a highly heritable, polygenic disease; heritability estimates for susceptibility to AS are more than 90%.1 ,2 Similarly there is also a major genetic contribution to disease severity in AS. The heritability of the commonly used disease severity metrics—Bath AS Disease Activity Index (BASDAI) and Bath AS Functional Index (BASFI)—have been estimated at 51% and 68%,3 respectively, and that of the Bath AS Radiological Index (BASRI) at 62%.4
To date, 34 loci have been identified that affect disease susceptibility to AS using case–control studies either with genome-wide genotyping microarrays5–7 or with the custom-designed Immunochip.8 There is substantial interest in identifying whether any of these genetic polymorphisms affect clinical or radiographic severity in AS. Only polymorphisms in the MHC and in ERAP1 have so far been reported in more than one study to affect clinical or radiographic severity.9–13 Several other studies have reported other genetic polymorphisms that correlate with disease or radiographic severity, but none have been replicated to date.
In this study, we have tested whether variants in genes involved in anabolic or catabolic bone pathways are associated with radiographic severity in AS. To measure radiographic severity, we used the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS),14 which provides an objective quantitative measure of radiographic change in patients with AS. It scores radiographic changes (erosion, sclerosis, squaring syndesmophytes) at 24 vertebral corners equally distributed between the cervical and lumbar spine. The mSASSS correlates moderately well with other disease severity measurements like the BASFI and can be used to predict BASFI.15
Patients and methods
Patients
All patients had definite AS according to the modified New York criteria.16 For the discovery phase, patients were recruited at one of seven clinics in Australia, UK and USA, participating in the Australo-Anglo-American Spondyloarthritis Consortium (TASC), and for the replication phase patients were recruited from two clinics in Canada and Australia, participating in the TASC or Spondyloarthritis Research Consortium of Canada (SPARCC). Written informed consent was obtained from all cases with approval from the relevant research ethics authorities at each participating centre.
Radiographic scoring
The mSASSS was used to assess radiographic severity in AS.14 Each radiograph used in the discovery and the replication phase was scored by one expert reader (MAB, TJL, MS, MMW, MHW, WPM and RDI). To assess the inter-reader variability, we selected 22 radiographs, a cross-sectional set from 10 patients and a longitudinal set of radiographs (including two time points) from each of six patients. We asked four of the readers (MAB, TJL, MMW and MHW) to score each set of radiographs. Longitudinal radiographs were scored blinded to time point. We estimated the inter-reader agreement using Fleiss’ κ statistic and pairwise mSASSS correlations. Inter-reader reliability was also assessed in two modified versions of the mSASSS to investigate whether inter-reader agreement improves with such modifications. These modifications removed squaring, sclerosis and erosion (score of 1) from the mSASSS, as assessing these features, particularly in the cervical spine, is unreliable and likely to contribute to variation in mSASSS among readers.17 Further, while the transition from non-bridging to bridging syndesmophytes is well established, whether squaring, sclerosis or erosions are precursors of non-bridging syndesmophytes is less well established. In version A, which we designated mSASSS_012, we collapsed classical mSASSS of 3 (denoting bridging syndesmophyte) to 2, scores of 2 (denoting presence of non-bridging syndesmophyte) to 1 and scores of 1 (denoting squaring, sclerosis or erosion) to 0. In version B, which we designated mSASSS_01, we collapsed classical mSASSS of 3 and 2 to 1 and scores of 1 to 0.
Genotyping
DNA was available from 688 cases who were scored using the mSASSS. Single nucleotide polymorphism (SNP) marker sets were designed to capture over 90% of the common haplotypic variation in the exons, exon–intron boundaries and 5 kb of the 5′ and 3′ UTR flanking 74 genes involved in anabolic or catabolic bone pathways. Genes were selected on the basis of their being key components of known bone anabolic or bone resorptive pathways, focusing on pathways identified in studies of AS itself (including studies in humans and mouse models), and of genes associated with variation in bone mineral density. Genes demonstrated to be associated with susceptibility to AS were also selected. This was based on the published literature (February 2009). Pairwise tagSNP selection was performed using HapMap Phase 218 and data analysed using Haploview,19 with a linkage disequilibrium threshold of 0.8. We selected 498 SNPs, which were genotyped using the Applied Biosystems OpenArray platform (Foster City, California, USA). In a second phase, 15 SNPs achieving p<0.05 were genotyped in a replication cohort comprising 830 patients from Canada and Australia, also using the Applied Biosystems OpenArray platform. Genotyping of HLA-B*27 was inferred from the tagSNP rs116488202 in the Illumina Immunochip platform20 as part of a case–control study described elsewhere.8 This SNP has >98% sensitivity and specificity for typing HLA-B*27.
Association analysis
Association analysis was performed by identifying patients with severe radiographic changes (cases) and comparing them with those who had mild radiographic changes (controls). For each patient, the most recent radiograph was analysed. In the event that either the cervical or the lumbar radiograph showed fusion in all vertebra (mSASSS of 36) the earlier radiograph without the maximum score was used. Radiographic scores were corrected for disease duration, gender and the interaction between disease duration and gender by linear regression. Residual values were computed from the fitted model, and subjects were dichotomised as having mild radiographic changes (lowest 40%) or severe radiographic changes (upmost 40%). This dichotomisation permitted the analysis of the data as a case–control study and the selection of patients with a more extreme phenotype while maximising the power of the study (see power calculation in ‘Methods’) and minimising the number of subjects excluded from the study (20% of radiographs with residual values around the median). Genotype associations were then performed with the allelic test (d.f. 1) in PLINK.21
For the replication phase, genotype and phenotype data were analysed as for the discovery phase. Results from both phases were then combined using fixed-effects meta-analysis.
Power calculation
We determined the power of our association study to identify genetic effects for radiographic changes by simulation. Power was estimated for a range of minor allele frequencies and effect sizes (assuming an additive effect), and from these simulations the optimal cut-off for dichotomising the data into subjects with severe and mild radiographic changes was determined.
For each simulation, we assumed a causal SNP with minor allele frequency between 0.02 and 0.50 with an additive genetic effect size ranging between 0.1 and 9.0 mSASSS units per minor allele, and a dichotomising cut-off threshold assigning the bottom or top 10, 20, 30 and 40% of the simulated films as having mild or severe radiographic changes, respectively. In each combination of allele frequency, effect size and dichotomising threshold, we simulated 1222 radiographic entries, where we independently sampled residuals, disease duration and gender from the total set of radiographs. Genotypes for the simulations were sampled from a Bernoulli random variable with allele frequency fixed for each simulation. For each simulated entry, we predicted the mSASSS with the regression model generated with the complete set of radiographs and generated an mSASSS with a genetic effect by adding the predicted score, the sampled residual and the genetic effect given by the additive effect size and the simulated genotyped for that entry. Simulated entries where by chance the mSASSS with the genetic effect had values below 0 or above 72 were removed from the simulations. Simulated entries were then dichotomised into subjects with severe radiographic changes and those with mild radiographic changes based on the specified thresholds. A contingency table was generated with the simulated genotypes and the dichotomised mSASSS, and an association test was performed with Fisher's exact test. The simulation was repeated 10 000 times for each combination of parameters, and power was determined by the proportion of times the p value for the association test was below significance (α=0.05).
Results
Characteristics of the study patients
A total of 2144 patients were enrolled and scored either by five different clinicians from the TASC (n=5) or by two investigators from the SPARCC consortium (table 1). The mean disease duration at baseline for the combined cohort was 20.2 years, which was greater in men than in women (20.6 vs 19.2 years; p=0.03, two-sided t test).
Demographic features of study cohorts at baseline*
Reliability analysis
Twenty-two radiographs were scored by four TASC readers, and comparison was performed with different metrics. Assessment of scores assigned to each vertebral corner demonstrated moderate agreement (κ>0.41) for all but T1 upper, where only slight agreement was observed (see online supplementary table S1). At this corner, we also observed the lowest rate of complete agreement between the four readers in the 22 radiographs and the largest proportion of radiographs where there was no agreement whether the site could be scored or not (in 8 out of 19 films at least one reader assigned a ‘not visualised’ score while at least one reader scored the site). Rates of complete agreement increased substantially from an average of 69.7% to 81.4% when collapsing the scores to mSASSS_012, but only marginally improved to an average of 83.5% when collapsing the scores to mSASSS_01.
Pairwise correlations between cervical, lumbar and total mSASSS were all found to be high between readers (≥0.90, see online supplementary table S2). Higher correlations were obtained for the lumbar spine than for the cervical spine, consistent with other reported comparisons. Collapsing the scores did not result in higher correlations between readers (data not shown).
The modified mSASSS were highly correlated with the mSASSS (r2>0.96) and associated with disease duration and with the disease severity measurement of spinal mobility Bath Ankylosing Spondylitis Metrology Index (r=0.79 with mSASSS_012 and mSASSS_01).
Radiographic severity is associated with clinical variables
Disease duration and gender, and its interaction, were found to be strongly associated with radiographic severity in AS (table 2), with radiographic damage progressing faster in men than in women (table 2, figure 1). For total mSASSS, among men the mean rise was 0.85 mSASSS units per year, while among women the mean rise was 0.45 mSASSS units per year.
Disease duration and gender are two clinical variables associated with radiographic severity in ankylosing spondylitis

Clinical features correlated with radiographic severity. Males are represented by filled triangles and females by filled circles. Lines depict the projection of the gender axis (dashed for males; dotted for females). Greyed samples were determined to be outliers (residual >3 SD).
Association analysis
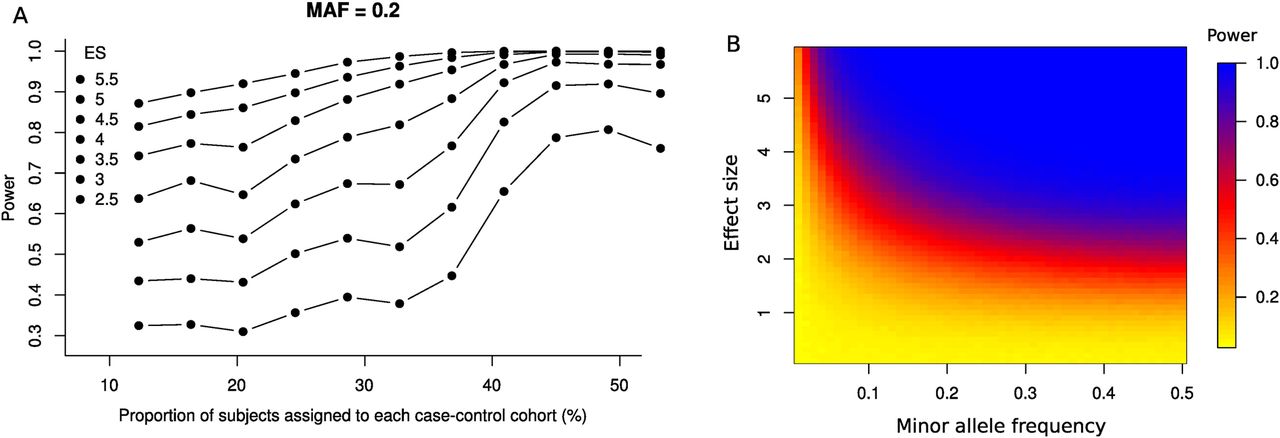
In total, 463 of 498 markers passed quality control filters (<10% missingness, Hardy–Weinberg equilibrium p <5×10−3); the mean call rate per SNP and sample was above 99%. As shown in figure 2, the statistical power to detect an association was maximal when score residuals were dichotomised into the lower 40% (‘mild’ radiographic change) and upper 40% (‘severe’ radiographic change). For markers with a minor allele frequency of 0.2 and significance threshold of p=0.05, the study had 80% power to detect a difference of 3.1 in mSASSS between these two arms.

Power calculations for genetic association tests. (A) Effect on power for different inclusion criteria in the dichotomisation of modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS). A genetic variant with a minor allele frequency (MAF) of 20% is assumed and several effect sizes (ES) are simulated, ranging from 2.5 to 5.5 mSASSS units per minor allele. (B) For an inclusion criteria of 40%, power is given for different combination of minor allele frequencies and simulated effect sizes. Effect sizes are disease duration and gender corrected.
Of the 15 SNPs carried forward into phase 2, 11 were successfully genotyped, 2 of which achieved replication of the discovery phase findings. The SNP rs8092336 achieved experiment-wide significance with the total mSASSS in phase 1 (p≤1×10−4; based on a Bonferroni correction for 463 independent tests) and was replicated in phase 2 (total mSASSS, preplication=0.02; pcombined 1.2×10−5, OR (95% CI)=0.31 (0.19 to 0.53)) (table 3; a complete list of results for all SNPs in the discovery phase is presented in online supplementary table S3). The minor allele of this SNP was found to have a moderate protective effect on radiographic severity in the cervical (pcombined=0.03) and lumbar (pcombined=0.02) mSASSS components. In the discovery cohort, patients with at least one copy of the minor allele had lower total mSASSS at baseline (p=0.05, single-sided t test, median difference in mSASSS of 3) (figure 3). This SNP represents a synonymous base change in the receptor activator of nuclear factor κ B gene (RANK), also known as tumour necrosis factor receptor superfamily, member 11a (TNFRSF11A).
Evidence of association with radiographic severity in ankylosing spondylitis in 11 SNPs genotyped in the discovery and replication phase

{kind=link}
{kind=link}
{kind=link}
Box plot of total modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) according to genotype for single nucleotide polymorphism (SNP) rs8092336, which shows association with total mSASSS (p value=1.2×10−5).
A further SNP, rs1236913, showed a protective association in phase 1 (p=0.04), phase 2 (p=3×10−3) and combined (OR (95% CI)=0.53 (0.35 to 0.80), p=2.6×10−3). This SNP lies in PTGS1, encoding Prostaglandin-Endoperoxide Synthase 1, also known as cyclooxygenase 1.
No association was observed between HLA-B*27 and cervical, lumbar or total mSASSS (p>0.05).
Discussion
Previous studies have shown that genetic variation is an important determinant of radiographic change in AS. Identifying the relevant polymorphisms and the biological pathways they influence could improve our understanding of the mechanisms of ankylosis in AS.
This is the first study systematically investigating genes involved in catabolic and anabolic bone pathways and their potential role in radiographic damage in AS. We observed experiment-wide significant association with rs8092336 in RANK, which encodes a TNF superfamily receptor, which regulates osteoclast activation22 and also interactions between T cells and dendritic cells.23 SNPs in RANK have previously been associated with osteoporosis,24 ,25 Paget's disease of bone26 and familial expansile osteolysis.27 Further studies will be required to determine whether the association observed here with mSASSS replicates in other AS cohorts, what the key associated variant(s) are and what functional relevance they have in AS. Our finding suggests that factors linking inflammation and bone resorption are important in the osteoproliferation in AS, with obvious potential therapeutic implications. This is consistent with previous in vitro data suggesting overactivity of RANKL-mediated osteoclastogenesis in AS.28
Association was also replicated in both phases with the SNP rs1236913, lying in PTGS1, a key enzyme in prostaglandin synthesis. Although this gene has not been previously associated with any common human disease, there is strong evidence for the involvement of prostaglandins in AS inflammation and osteoproliferative disease. The gene PTGER4, encoding the prostaglandin E2EP4 receptor, is associated with AS.6 ,8 Inhibition of cyclooxygenase enzymes with non-steroidal anti-inflammatory drugs is highly effective in treating pain in AS, and there is some evidence from observational and controlled trials suggesting that NSAID treatment may retard the progression of radiographic change in AS.29–32 These findings require further replication, but they do lend some support to the growing evidence of involvement of prostaglandin pathways in AS-associated osteoproliferation.
We observed no association between HLA-B*27 and radiographic severity in this study, confirming our previous reports.33 This is also consistent with studies demonstrating no difference in disease activity or functional impairment measures in HLA-B*27-positive and HLA-B*27-negative cases.34 Thus, while HLA-B*27 is clearly associated with AS susceptibility, it is not associated with the severity of ankylosis in the condition. A recently published analysis of the OASIS cohort suggests that HLA-B*27 and male gender may be associated with more rapid radiographic progression in AS, but this was a small study of only 186 cases.35 The subgroup analyses in this study were based on only ∼32 HLA-B*27-negative and ∼56 female cases, so these conclusions should be treated with caution. However, we also noted that men had more severe disease than women.
All studies to date investigating the role of genetic polymorphisms in disease severity and radiographic change in AS have been significantly underpowered (n<500) because of the effort and cost required to gather large cohorts with the necessary phenotypic data. This study, despite being the largest to date performed in AS (in total 2144 AS cases), still does not have adequate power to exclude small-medium genetic effects on radiographic severity. Further, it was not truly systematic since it did not tag all the variants in the genes studied and was cross-sectional and not longitudinal. In cross-sectional studies, adjustment for disease duration depends on case recall of the age of symptom onset, whereas in longitudinal studies the interval between observations is known precisely.
In this study, we have used mSASSS, a well-established metric of disease severity in AS, as a quantitative measurement of radiographic change. Our results demonstrate that with well-trained personnel inter-reader correlation for mSASSS can be high; this is important for the study of big cohorts with the large quantities of phenotypic data required for genetic studies. We also demonstrate that small modifications can be made to scoring mSASSS to reduce the inter-reader variability; these include collapsing the scores and removing the upper corner of the T1 vertebra from the analysis as it is difficult to visualise radiographically.
We have demonstrated that this type of study can be effective at demonstrating genetic effects involved in aspects of disease severity in AS, but more powerful and comprehensive studies will be required to identify the full complement of genes involved. This could in time provide very useful insights into the osteoproliferative processes in AS, potentially assisting the identification of drug targets to slow down radiographic progression not targeted by current treatments.
Acknowledgments
We would like to thank all the participants who provided the case and control DNA and clinical information necessary for this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
AC, WPM, MAB and MHW contributed equally.
Correction notice This article has been corrected since it was published Online First. The author ‘Lianne S Gensler’ has been added.
Contributors AC and MAB performed statistical analysis and wrote the manuscript. WPM, RDI, MAS, MMW, TJL, MAB and MW read and scored radiographs. PD performed genotyping. BPW contributed to editing of the manuscript. MAB conceived the study. All other authors contributed with subject recruitment and study design, and approved the manuscript.
Funding This study was funded by the US National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grants P01-052915. MMW is supported by the Intramural Research Program, NIAMS/NIH. MAB was funded by a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship. Recruitment of patients to this study was in part funded by a project grant from Arthritis Research UK (19536). WPM is a medical scientist of Alberta Innovates Health Solutions.
Competing interests None.
Patient consent Obtained.
Ethics approval University of Queensland and Princess Alexandra Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.