Article Text

Abstract
Objectives Randomised, double-blind, placebo-controlled study to evaluate efficacy and safety of tabalumab in patients with rheumatoid arthritis (RA) with inadequate responses to methotrexate (MTX-IR).
Methods 1041 patients with moderate–severe RA despite ongoing MTX enrolled in a 52-week study evaluating subcutaneous tabalumab 120 mg every four weeks (120/Q4W) or 90 mg every two weeks (90/Q2W) versus placebo. Primary endpoints were American College of Rheumatology 20% (ACR20) response rate and Health Assessment Questionnaire-Disability Index change from baseline at 24 weeks and modified Total Sharp Score (mTSS) change at 52 weeks.
Results There were no significant differences in ACR20 responses at week 24 or mTSS change from baseline at week 52 among treatment groups. Declines were seen in CD20+ B cells and immunoglobulin levels in tabalumab groups, but not placebo: B cells (−15.0%, −18.8%, 5.3%, in the 120/Q4W, 90/Q2W, and placebo groups, respectively); IgM (−16.3%, −19.4%, −0.1%), IgA (−11.4%, −4.7%, 1.2%) and IgG (−8.6%, −7.8%, 0.1%). Discontinuations due to adverse events were similar between groups. Numerically more serious infections were reported in tabalumab groups (1.7%, 0.6%, 0.3%); numerically more injection-site reactions were reported in the 90/Q2W group (2.3%, 4.3%, 2.3%).
Conclusions Neither clinical efficacy nor significant safety signals were observed with tabalumab despite evidence of biological activity. This study was terminated early due to insufficient efficacy.
Trial registration number NCT01198002.
- B cells
- Autoimmune Diseases
- Rheumatoid Arthritis
Statistics from Altmetric.com
Introduction
B-cell-activating factor (BAFF) is increased in patients with rheumatoid arthritis (RA). Therefore, neutralising BAFF may be an effective method of targeting B cells and treating RA. However, agents targeting BAFF including belimumab1 and atacicept2 ,3 have shown disappointing efficacy in RA. It is unclear whether this inadequate response is a target-specific or compound-specific issue. Tabalumab, a fully human anti-BAFF monoclonal antibody that binds and neutralises both soluble and membrane-bound BAFF,4 reduced signs and symptoms of RA by up to 38% compared with placebo in phase II clinical trials of patients with active RA also taking methotrexate (MTX) treatment.5 ,6 The phase III study reported herein evaluated the efficacy and safety of tabalumab in RA.
Methods
This multicentre, randomised, double-blind, placebo-controlled, phase III study (NCT01198002) evaluated the efficacy and safety of tabalumab 120 mg every four weeks (120/Q4W) or 90 mg every two weeks (90/Q2W) compared with placebo in patients on background MTX with moderate–severe RA and inadequate responses to MTX (MTX-IR). Patients were enrolled from 23 countries in North America, Central and South America, Europe, Asia and Australia. The tabalumab doses were chosen based on data from phase II RA studies and pharmacokinetic modelling and simulations.5 ,6
Patient population
Adults (≥18 years) with RA (≥6 months' duration) with ≥8/68 tender and 8/66 swollen joints, positive rheumatoid factor (RF) and/or anticyclic citrullinated peptide antibody, regular use of MTX for ≥12 weeks with stable doses (10–25 mg/week) for ≥8 weeks before baseline, ≥1 RA-related joint erosion on hand or foot X-rays by central reading and screening C-reactive protein (CRP) >1.2 times the upper limit of normal or screening erythrocyte sedimentation rate >28 mm/h were eligible for enrolment. Patients who received any biological disease-modifying antirheumatic drug with discontinuation due to insufficient efficacy were excluded (see online supplementary text for more detailed information).
Study design
Patients were randomised 1:1:1 to receive subcutaneous (SC) tabalumab 120/Q4W (240 mg SC loading dose, given as two SC injections of 120 mg), 90/Q2W (180 mg SC loading dose, given as two SC injections of 90 mg) or placebo (two SC placebo injections). Non-responders at week 16 (<20% improvement from baseline in both tender and swollen joint counts) were assigned to receive tabalumab 90/Q2W for the remainder of the study and were unblinded at week 24. Results through week 52 are presented here. An open-label treatment period (weeks 52–100) was planned but was interrupted due to early termination of the study; these data will not be discussed.
Objectives
The primary objective was to evaluate the efficacy of tabalumab 120/Q4W or 90/Q2W compared with placebo in patients with RA (all on background MTX) as measured by the co-primary endpoints of the American College of Rheumatology 20% (ACR20) response rates and change from baseline in the Health Assessment Questionnaire-Disability Index (HAQ-DI) scores at week 24 and structural progression on the van der Heijde modified Total Sharp Score (mTSS)7 at week 52.
Statistical analyses
Significance testing for the primary analysis of ACR20 at week 24 used the Dunnett procedure at overall two-sided significance levels of 0.05 with each tabalumab dose versus placebo carried out at a two-sided alpha level of 0.0271. Efficacy analyses were conducted on the efficacy population (all randomised patients, excluding patients with low MTX dose (<10 mg/week)); safety analyses were conducted on the safety population (randomised patients who received ≥1 dose of study drug). A logistic regression model was used for ACR20 adjusting for treatment, geographic region and baseline Disease Activity Score in 28 Joints–CRP; an analysis of covariance model (ANCOVA), adjusted for treatment, geographic region and baseline value, was used for change from baseline HAQ-DI and mTSS. Fisher's exact test for categorical data and analysis of variance for continuous data were used unless otherwise specified.
For the ACR20, ACR50 and ACR70 response analyses, all non-responders at week 16 and patients who discontinued early were defined as non-responders (non-responder imputation) from this visit onward. For HAQ-DI and mTSS, modified baseline-observations-carried-forward (mBOCF) and linear extrapolation methods were used to impute missing data, respectively; other continuous variables on safety were analysed using ANCOVA with a modified last observation carried forward (mLOCF) used to impute missing data. In mLOCF, for all non-responders assigned to 90/Q2W at week 16, the last non-missing observation at or before week 16 was carried forward to subsequent time points for evaluation. For all other patients discontinuing study treatment for any reason, the last non-missing observation before discontinuation was carried forward to subsequent time points for evaluation. mBOCF is identical to mLOCF with one exception: for patients discontinuing study treatment due to an adverse event (AE), the baseline observation was carried forward to all postbaseline time points for evaluation. A preplanned futility analysis was conducted after approximately 50% of the patients had completed week 24.
Results
Patient disposition
In total, 1041 patients were randomised to treatment (intention-to-treat (ITT) population) (see online supplementary figure S1); 587 patients (56.4%), excluding week 16 non-responders, completed to week 24 (primary endpoint for ACR20 and HAQ-DI) and 177 patients (17.0%) completed to week 52 (primary endpoint for mTSS). Three randomised patients (90/Q2W=2; placebo=1) did not receive study drug, resulting in a safety population of 1038 patients. Also, 210 patients (20.2%) were non-responders at week 16; 654 patients (62.8%) discontinued treatment before reaching week 52. The most common reason for discontinuing treatment was sponsor decision to terminate the study (515/1041, 49.5%); 369/515 (71.7%) patients who discontinued due to sponsor decision did so after week 24 (these numbers exclude non-responders after week 16).
Baseline characteristics
Demographic and baseline disease characteristics were similar between all groups for the ITT population (see online supplementary table S1).
Clinical response
The percentage of patients achieving ACR20 at week 24 (primary endpoint) was 29.7%, 32.8% and 25.1% in the 120/Q4W, 90/Q2W and placebo groups, respectively. There were no significant differences between each tabalumab group and placebo group in the percentage of patients achieving ACR20, ACR50 or ACR70 at week 24 (figure 1). There was some variability between geographic regions, but there were no significant treatment-by-subgroup interactions (p=0.738) for ACR20 at week 24 (see online supplementary table S2).Similarly, there were no clinically meaningful changes from baseline to week 24 in HAQ-DI scores: least-squared means (SE) were −0.26 (0.03) for 120/Q4W, −0.3 (0.03) for 90/Q2W and −0.2 (0.03) for the placebo group; p=0.132 and p=0.007 versus placebo, respectively, with similar baseline scores. There were no significant changes from baseline to week 52 in observed mTSS: least-squared means (SE) were 1.43 (0.27) for 120/Q4W, 0.84 (0.27) for 90/Q2W and 1.57 (0.27) for the placebo group; p=0.704 and p=0.052, respectively (cumulative probability plot shown in online supplementary figure S2).

American College of Rheumatology Responder Index (ACR) response at week 24. p Value for 120 mg once every four weeks (120/Q4W) versus placebo=0.246; p value for 90 mg once every two weeks (90/Q2W) versus placebo=0.029.
Biological activity
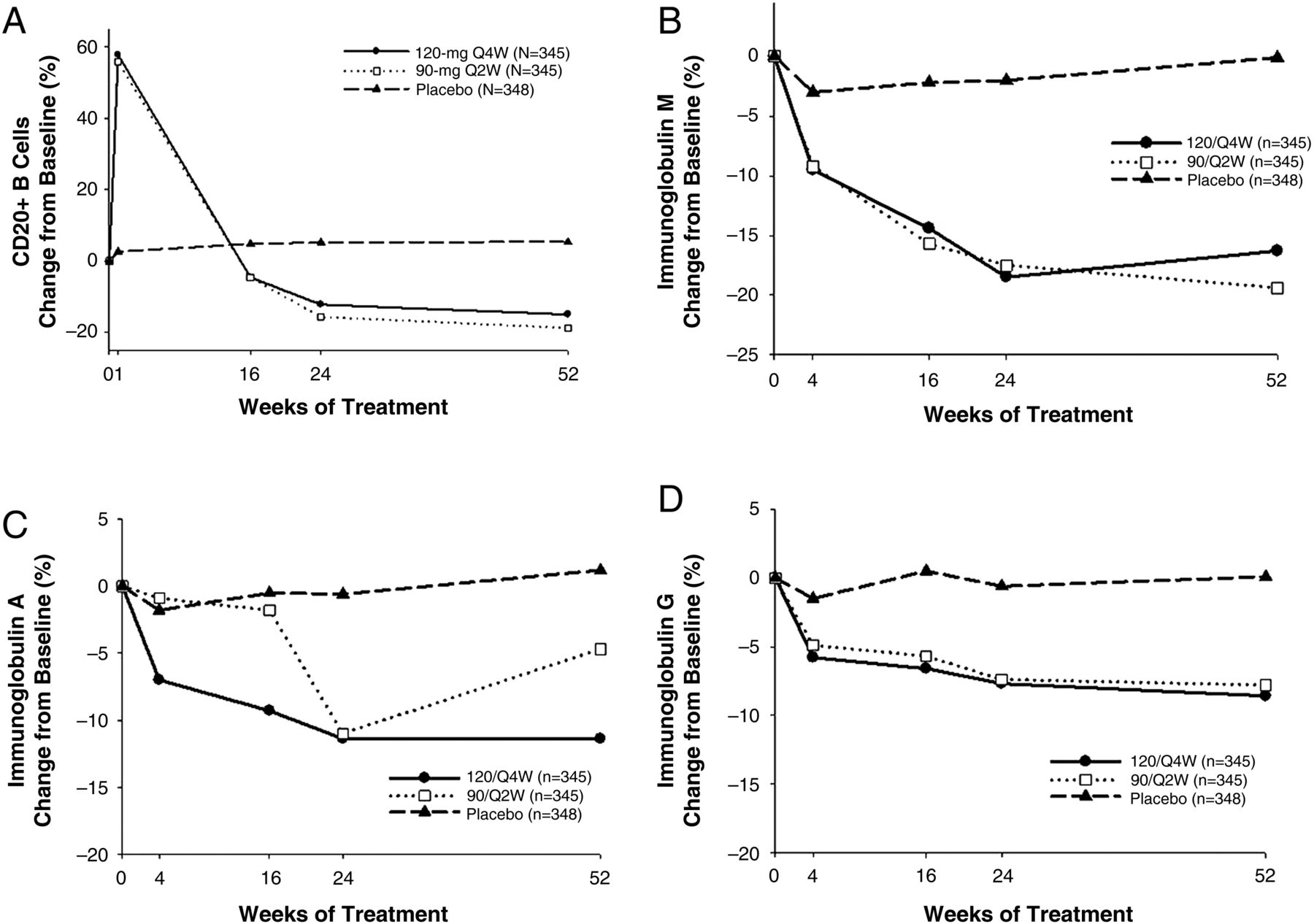
An initial increase in median CD20+ B-cell counts occurred in tabalumab-treated patients at week 1 (mean per cent change from baseline: 120/Q4W: +57.8%; 90/Q2W: +55.9%) followed by a subsequent decrease from week 4 to week 52 (mean per cent change from baseline: 120/Q4W: −15.0%; 90/Q2W: −18.8%) (figure 2A). There were significant differences in absolute and relative (%) CD20+ B-cell-level changes from baseline to week 52 mLOCF endpoints in both tabalumab groups compared with the placebo group (p<0.001). There were also significant decreases in RF levels from baseline to week 52 mLOCF (see online supplementary table S3) and in immunoglobulin (Ig) levels at week 52 in the tabalumab groups (figure 2B–D); changes from baseline were also significantly greater for the tabalumab groups compared with the placebo group (p<0.001 for all comparisons).

{kind=link}
{kind=link}
Mean per cent change in total (CD20+) B cells over 52 weeks (A) and mean per cent change for IgM (B), IgA (C) and IgG (D); values are observed for baseline through week 24 and are modified last observation carried forward for week 52. 120/Q4W, 120 mg once every four weeks; 90/Q2W, 90 mg once every two weeks.
Safety
Frequencies of treatment-emergent adverse events (TEAEs) were similar between all three groups as were discontinuations due to AEs, TEAEs and serious AEs (see online supplementary table S4). The 90/Q2W group had a higher percentage of injection-site reactions (4.3%) compared with the 120/Q4W (2.3%) and placebo (2.3%) groups. Additionally, although the percentage of patients who received tabalumab and reported at least one treatment-emergent infection (120/Q4W: 24.1%; 90/Q2W: 22.6%) was similar with the placebo group (26.1%), there was a numerically higher frequency of serious infectious events in the 120/Q4W (1.7%) group compared with the 90/Q2W (0.6%) and placebo (0.3%) groups. Three patients died: two in the 120/Q4W group (sepsis and myocardial infarction) and one in the placebo group (myocardial infarction). The appearance of anti-tabalumab antibodies was not associated with any clinical or safety signals; all antibodies were non-neutralising.
Discussion
Previous assessments of tabalumab in patients with RA who were MTX-IR showed improvement in clinical features in controlled phase II5 ,6 and open-label studies.8 This phase III study of tabalumab in patients with RA with MTX-IR did not meet its primary endpoint of ACR20 or HAQ-DI superiority over placebo at 24 weeks or mTSS differences at 52 weeks.
Despite clear pharmacodynamics changes as evidenced by changes in CD20+ B cells, RF and immunoglobulins following tabalumab treatment, BAFF inhibition with tabalumab was not clinically, functionally or structurally efficacious in patients with moderate–severe RA taking MTX. Given that the placebo response in this study was as expected for an MTX-IR population, the absence of a significant effect of active treatment was not due to a higher-than-anticipated placebo response. The safety results of tabalumab were similar to those observed in previous studies4 ,5 with the exception of numerically more serious infectious events (120/Q2W) and more injection-site reactions (90/Q2W) compared with the placebo group.
Based on findings in this study and data from a preplanned futility analysis, Eli Lilly and Company discontinued the tabalumab RA programme due to lack of efficacy; the decision was not based on safety concerns. Data from smaller phase II trials may not be sufficiently reliable to foresee the outcomes of larger phase III RA clinical trials. In the future, it will be important to evaluate factors in small phase II trials that may predict failure in phase III.
BAFF blockade with tabalumab has unique effects on the distribution of human B-cell subsets in the bloodstream. BAFF blockade causes increases in blood levels of memory B cells, which after several months return to pretreatment levels.9 Patterns similar to tabalumab's have been seen with BAFF blockers belimumab1 and atacicept.2 ,3
Although clinical efficacy was seen in the phase II studies of tabalumab in the MTX-IR population and despite pharmacodynamics evidence that the drug had biological activity, the results of this larger phase III study show that tabalumab does not provide the level of clinical benefit in patients with RA who are MTX-IR seen with other currently approved biological agents. Hence, BAFF blockade alone does not appear to provide sufficient interference with the immunopathogenesis of RA. No unexpected safety signals were seen with the drug. As a result of a lack of efficacy, the study was terminated early, but most patients completed 24 weeks, thus there were sufficient data to analyse the effect of tabalumab on the primary endpoints of ACR20 and HAQ-DI.
Acknowledgments
We would like to thank Pamela W Anderson, MD of Eli Lilly and Company (Indianapolis, Indiana, USA), and Kathy Oneacre, MA, of inVentiv Health Clinical (Princeton, New Jersey, USA), for assistance with the development of this manuscript. We would also like to thank all of the study investigators for their contribution to the study.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table S1
- Data supplement 2 - Online table S2
- Data supplement 3 - Online table S3
- Data supplement 4 - Online table S4
- Data supplement 5 - Online supplement
- Data supplement 6 - Online figures
Footnotes
Handling editor Tore K Kvien
Contributors Each author has participated directly with the research and drafting of the article.
Funding This study was sponsored by Eli Lilly and Company and/or one of its wholly owned subsidiaries.
Competing interests JSS has received research support from Pfizer and has been a consultant for Pfizer and Eli Lilly and Company; MEW has been a consultant for Eli Lilly and Company, Pfizer, and VertexD; DvdH has been a consultant for Eli Lilly and Company, AbbVie, Amgen, AstraZeneca, Augurex, BMS, Celgene, Centocor, Chugai, Covagen, Daiichi Sankyo Ltd, GlaxoSmithKline, Janssen Biologics, Merck, Novartis, Novo-Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB, and Vertex; WFCR has been a consultant for Eli Lilly and Company; RvV received research support from AbbVie, BMS, GSK, Pfizer, Roche, UCB and has been a consultant for AbbVie, Biotest, BMS, GSK, Janssen, Eli Lilly and Company, Merck, Pfizer, Roche, UCB and Vertex; COB has received research support and has been a consultant for Eli Lilly and Company; MV, AG, FZ, WJK, RO and CL are shareholders of and employees of Eli Lilly and Company; P-YB is an employee of Eli Lilly and Company.
Patient consent Obtained.
Ethics approval The study protocol was approved by local institutional review boards in accordance with the Declaration of Helsinki and applicable laws and regulations.
Provenance and peer review Not commissioned; externally peer reviewed.