Article Text

Abstract
Objectives Bruton's tyrosine kinase (Btk) is required for B lymphocyte and myeloid cell contributions to pathology in murine models of arthritis. Here, we examined the potential contributions of synovial Btk expression and activation to inflammation in rheumatoid arthritis (RA).
Materials and methods Btk was detected by immunohistochemistry and digital image analysis in synovial tissue from biologically naive RA (n=16) and psoriatic arthritis (PsA) (n=12) patients. Cell populations expressing Btk were identified by immunofluorescent double labelling confocal microscopy, quantitative (q-) PCR and immunoblotting. The effects of a Btk-specific inhibitor, RN486, on gene expression in human macrophages and RA synovial tissue explants (n=8) were assessed by qPCR, ELISA and single-plex assays.
Results Btk was expressed at equivalent levels in RA and PsA synovial tissue, restricted to B lymphocytes, monocytes, macrophages and mast cells. RN486 significantly inhibited macrophage IL-6 production induced by Fc receptor and CD40 ligation. RN486 also reduced mRNA expression of overlapping gene sets induced by IgG, CD40 ligand (CD40L) and RA synovial fluid, and significantly suppressed macrophage production of CD40L-induced IL-8, TNF, MMP-1 and MMP-10, LPS-induced MMP-1, MMP-7 and MMP-10 production, and spontaneous production of IL-6, PDGF, CXCL-9 and MMP-1 by RA synovial explants.
Conclusions Btk is expressed equivalently in RA and PsA synovial tissue, primarily in macrophages. Btk activity is needed to drive macrophage activation in response to multiple agonists relevant to inflammatory arthritis, and promotes RA synovial tissue cytokine and MMP production. Pharmacological targeting of Btk may be of therapeutic benefit in the treatment of RA and other inflammatory diseases.
- Rheumatoid Arthritis
- Inflammation
- Cytokines
- Chemokines
Statistics from Altmetric.com
Introduction
Recent studies have identified a potentially important role for phosphatidylinositol 3-kinase (PI3K) signalling in maintaining cellular recruitment, activation and survival in rheumatoid arthritis (RA) synovial tissue.1 The primary function of PI3K family members is to provide membrane targeting signals which mediate recruitment of selected proteins to the plasma membrane. PI3Ks catalyse the phosphorylation of phosphatidylinositol (PI) to generate the second messengers PI(3) P, PI(3,4) P2, PI(3,5) P2 and PI(3,4,5) P3, which recruit and regulate the function of pleckstrin homology (PH) domain-containing proteins.1 Genetic deletion of either the γ or δ isoforms of the PI3K p110 catalytic subunit, or treatment with specific inhibitors of these PI3K isoforms, has been shown to be protective in murine models of arthritis, associated with reduced chemotactic responses, cytokine production and survival of immune cells.2–4
Protein kinase B (PKB) and Tec family kinases represent two downstream targets of PI3 K signalling which have been best characterised within the context of RA.1 PKB is a serine/threonine kinase which regulates mammalian target of rapamycin (mTOR) and forkhead box O (FoxO) transcription factors. Levels of activated, phosphorylated (p-) PKB are elevated in RA versus non-RA synovial tissue, and PKB-dependent phosphorylation of FoxO4 in synovial macrophages, particularly in the synovial sublining, is significantly enhanced in RA.5 ,6 Tec family kinases include Tec, Bruton's tyrosine kinase (Btk), Itk, Bmx and Rlk.7 Complete activation of Tec family kinases upon cell-surface receptor triggering requires PI3K-dependent relocalisation of the protein to the plasma membrane, subsequent phosphorylation by a Src family kinase, and finally autophosphorylation.8 In addition to the well-studied role of Tec family kinases in PLCγ activation and Ca2+ mobilisation, they can also act downstream of numerous cell-surface receptors that influence a wide range of signalling pathways involved in cellular proliferation, differentiation, apoptosis, cell migration, and inflammatory gene transcription.7
Inactivating mutations in the gene encoding Btk cause X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (XID) in mice. Btk is absolutely required for human B cell development and function, and XLA is characterised by the complete absence of circulating B cells due to a block between pro- and pre-B cell stages, and a severe reduction in serum immunoglobulin levels.9 A similar, although milder syndrome is observed in XID mice.10 Apart from its role in B cells, where Btk mediates antigen receptor, CD40 and chemokine receptor signalling responses, Btk has also been shown to regulate activation of monocytes, macrophages, mast cells and platelets, and osteoclast differentiation.11–17 Btk deficiency in myeloid lineages leads to poor inflammatory responses, and XID mice develop less severe experimental autoimmune encephalitis, DSS-induced colitis, and carrageenan-induced acute oedema.18
A potential role for Btk in RA was first suggested by the finding that XID mice are resistant to collagen induced arthritis (CIA).19 Subsequent pharmacological development of specific Btk inhibitors and their assessment in experimental rodent models of RA has demonstrated that Btk activity in both B cell and myeloid cell populations contributes to disease.20–25 However, while this preclinical evidence suggests a therapeutic potential in targeting Btk in RA, little is known about the expression or function of Btk in RA synovial tissue. Here, we examined the expression of Btk in RA and psoriatic arthritis (PsA) synovial tissue, and the effects of a specific Btk inhibitor on gene expression in human macrophages and RA synovial explant cultures.
Materials and methods
Patients, immunohistochemical analysis and confocal microscopy
Synovial biopsies were obtained by needle arthroscopy as previously described from clinically active joints of RA (n=16) and PsA (n=12) patients.26 RA and PsA patients fulfilled the 1987 American College of Rheumatology criteria for RA and the CASPAR classification criteria for PsA.27 ,28 None of the patients had been previously treated with biologicals. Patient clinical characteristics are detailed in table 1. Synovial biopsies were also obtained from a second independent cohort of seven RA patients and seven osteoarthritis (OA) patients, as well as 8 additional RA patients for synovial explant studies (see online supplementary tables S1 and S2 for patient clinical characteristics). All patients gave their written informed consent prior to study inclusion, and this study was approved by the Medical Ethics Committee of the Academic Medical Center, University of Amsterdam. Immunohistochemical analysis, as previously described, and confocal microscopy techniques are detailed in the online supplementary Materials and methods.29 ,30
Clinical features of RA and PsA patients included in the study
Cell isolation, tissue culture, adenoviral transduction, measurement of cytokine production, measurement of mRNA expression and gene expression profiling
Detailed protocols of the isolation, culture and differentiation of lymphocyte, myeloid and FLS cells, adenoviral transduction of macrophages, measurement of cytokine production, RNA extraction, cDNA synthesis, quantitative PCR and gene expression profiling are provided in the online supplementary Materials and methods.
Macrophage stimulation
Cell culture grade Anti-Biotin MACSiBead Particles (Miltenyi Biotec) were loaded with biotinylated IgG1 (BioLegend) according to the manufacturer's instructions (30 µg biotinylated primary antibody per 1×108 bead particles). Macrophages were treated with either vehicle control or RN486 (1 µM) for 1 h, and then either left unstimulated or stimulated with IgG beads (1:1 bead:cell ratio), CD40L (1 µg/mL, R&D Systems), TNF (10 ng/mL, BioSource), LPS (10–1000 ng/mL, Sigma-Aldrich) or 10% RA patient synovial fluid (RA SF, pooled from five RA patients) for 4–24 h.
Statistical analysis
Data are presented as mean±SEM unless otherwise indicated. The Mann–Whitney U test was used for comparing synovial Btk expression between diagnostic groups. The Spearman's correlation coefficient was used for correlation analyses. Analyte concentration data were analysed using one- and two-sided Student t tests, as appropriate. Statistical significance was set at p<0.05.
Results
Btk is expressed in the synovial tissue of patients with inflammatory arthritis
We first examined the expression of Btk in RA and PsA synovial tissue. Specific staining of antibodies recognising Btk was observed in RA synovial tissue, while no staining was observed with irrelevant control antibodies (figure 1A). In both RA and PsA synovial tissue, Btk was most readily apparent in cellular infiltrates of the synovial sublining (figure 1B). Equivalent expression levels of Btk were observed in RA and PsA synovial tissue, as detected by digital image analysis (figure 1C). Similar levels of Btk expression were also observed in an independent analysis of RA and OA patients (figure 1D). No relationship was observed between Btk expression levels and patient clinical characteristics (serum C-reactive protein levels, erythrocyte sedimentation rate, DAS28 score, and ACPA- or RF-positivity) (data not shown). To examine the relationship between Btk expression and synovial cellular composition, sections were stained with antibodies against specific markers for T cells, B cells, FLS, macrophages, endothelial cells and mast cells. We found a positive correlation between Btk expression levels and numbers of CD68+ synovial macrophages (R=0.6324, p<0.01), CD163+ macrophages (R=0.7275, p<0.005) and CD3+ T cells (R=0.7903, p<0.001) (figure 1E), but no correlation with CD22+ B cells (figure 1E), CD55+ FLS, or vWF+ endothelial cells in RA synovial tissue (see online supplementary table S3). In PsA synovial tissue we did not observe any significant correlations with the expression of cellular markers (figure 1E and see online supplementary table S3), although all cellular markers were detected at similar levels in RA and PsA synovial tissue (see online supplementary table S4). These data demonstrate that Btk is expressed in the synovial tissue of arthritis patients, and suggest that at least in RA, synovial Btk expression correlates with myeloid and T cell infiltration.

Btk expression in rheumatoid arthritis (RA), psoriatic arthritis (PsA) and osteoarthritis synovial tissue. Tissue sections from patients with RA and PsA were stained with irrelevant control antibodies or with antibodies against Btk, CD3, CD22 and CD68. (A) Representative staining of RA synovial tissue with Btk and control (ctrl) antibodies (magnification upper panels ×200, bottom panels ×400). (B) Representative stainings of RA and PsA synovial tissue with Btk antibodies (magnification ×200). (C) Quantitative analysis of Btk expression in RA (n=16) and PsA (n=12) synovial tissue. (D) Quantitative analysis of Btk expression in RA (n=7) and osteoarthritis (OA, n=7) patient synovial tissue. Values are expressed as integrated optical density (IOD) per mm2 per number of nuclei calculated by computer-assisted digital image analysis. Data are presented as boxplots where the boxes represent the 25th and 75th percentiles, the line within the box denotes the median value, and the lines outside the box mark the 10th and 90th percentiles. (E) Correlation of Btk expression and number of cells/mm2 expressing CD22, CD68, CD163 and CD3 in RA (upper panels) and PsA (lower panels) synovial tissue. Correlations were calculated using Spearman's rank correlation coefficient. Circles indicate individual patient values, and Spearman R and p values are indicated in each graph. NS, not significant.
Btk is expressed in RA synovial myeloid cells and B lymphocytes
Immunofluorescent double labelling confocal microscopy was performed to identify which cell types express Btk in RA synovial tissue. Btk expression colocalised with CD22+ B cells, CD68+ macrophages and MCT+ mast cells (Spearman colocalisation coefficient 0.461, 0.674 and 0.506, respectively), but not CD3+ T cells, vWF+ endothelial cells, or CD55+ FLS (figure 2A and see online supplementary figure S1). Btk also colocalised with CD68+ and CD163+ macrophages in PsA synovial tissue (see online supplementary figure S2). Immunoblotting experiments performed on lysates from purified B cells, T cells, monocytes and GM-CSF-differentiated macrophages derived from healthy donor peripheral blood, as well as RA FLS, confirmed that Btk expression was restricted to B cells and myeloid cells (figure 2B). Itk protein expression was restricted to T cells, and Bmx protein was detected in all cell populations except for RA FLS (figure 2C). Similar results were obtained when examining the mRNA expression of Btk, with equivalent levels of Btk mRNA observed in both freshly isolated monocytes and differentiated macrophages (figure 2D). Itk was detected in T cells, while Bmx mRNA was detected at low levels in all cell types compared to positive control K562 cells (figure 2D), possibly indicating Bmx antibody cross-reactivity with other Tec family kinases (figure 2C). Similar results, as well as Btk expression levels, were observed following analysis of RA and PsA peripheral blood-derived macrophages differentiated in GM-CSF, and healthy donor macrophages differentiated in either 10% RA or 10% PsA SF (see online supplementary figure S3).

Btk expression in specific cell types in rheumatoid arthritis (RA) synovial tissue. (A) Representative immunofluorescent double-staining of RA synovial tissue with anti-Btk antibody (red), CD3, CD22, CD68 and vWF cell-specific markers (green) and DAPI (blue) (magnification ×400). (B) Protein extracts were prepared from healthy donor peripheral blood B cells, T cells, buffy coat monocytes (monos) and GM-CSF-differentiated macrophages (MΦ), RA FLS and K562 cells, and analysed by immunoblotting with antibodies recognising (B) Btk and tubulin or (C) Itk, Bmx and tubulin. Results are representative of three independent experiments. (D) Total RNA was extracted from healthy donor peripheral blood B cells, T cells, buffy coat monocytes (monos) and GM-CSF-differentiated macrophages (MΦ) (n=4 donors for each), RA FLS (n=3) and K562 cells, reverse transcribed and expression of Btk, Itk and Bmx mRNA was monitored by qPCR. Data are presented as mean±SEM expression relative to GAPDH, arbitrary units (a.u.).
Inhibition of Btk selectively prevents inflammatory mediator induction by macrophage agonists
To determine the potential contributions of macrophage Btk activation to inflammation in RA, we stimulated macrophages with IgG, CD40L, TNF or LPS, all agonists relevant to macrophage activation in RA. Of these, Btk involvement has previously been reported in B cell CD40 signalling, and monocyte Fc receptor and TLR signalling. Experiments were performed in the absence or presence of saturating concentrations of a specific Btk inhibitor, RN486.22 In the absence of stimulating agonists, neither carrier solvent (DMSO) nor RN486 had any effect on basal production of IL-6 (figure 3A) or other secreted products tested (see below) compared to macrophages in medium alone. In parallel, we compared the effects of carrier solvent and RN486 on agonist-induced IL-6 production. IgG-coated beads, CD40L, TNF and LPS (figure 3A) all induced IL-6 production in human macrophages derived from healthy donor (n=6) monocytes, as determined by ELISA. In the presence of RN486, macrophage IL-6 production in response to Fc receptor (40% inhibition, p<0.01) and CD40 stimulation (50%, p<0.05), but not TNF, was significantly inhibited (figure 3A). RN486 failed to influence LPS-induced IL-6 production at any of the LPS concentrations tested (figure 3A). CD40L, TNF and LPS all induced macrophage IL-8 production (figure 3B), but only IL-8 produced in response to CD40L was blocked in the presence of RN486 (p<0.01) (figure 3B). Macrophage transduction with adenoviruses encoding Btk, but not GFP, increased cellular expression of Btk (figure 3C) and increased macrophage IL-6 production in response to IgG (p<0.05) and CD40L stimulation (p<0.05) (figure 3D).
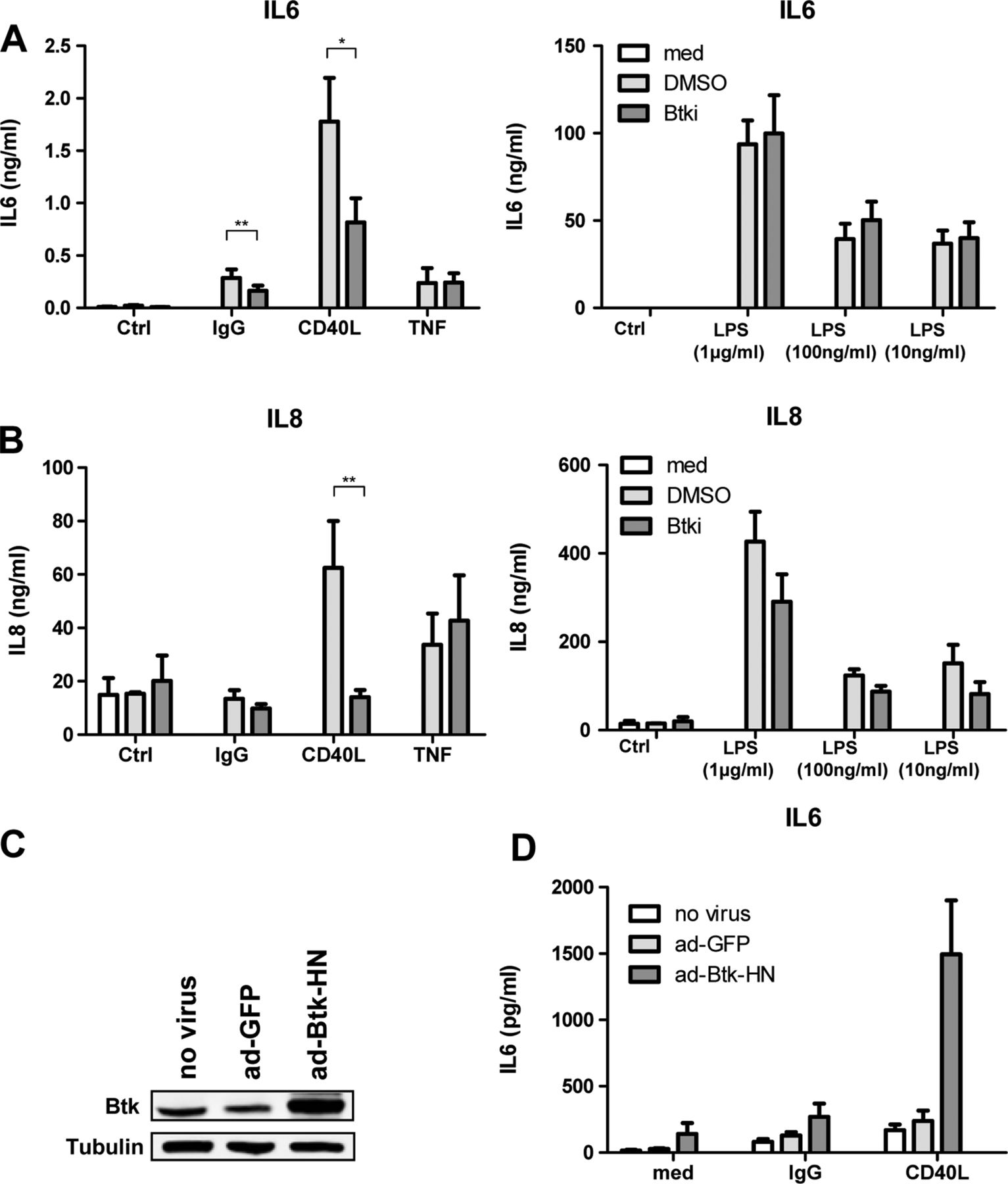
Btk inhibitor RN486 suppresses macrophage production of cytokines in response to IgG and CD40L stimulation. Analysis of (A) IL-6 and (B) IL-8 production in supernatants of macrophages stimulated with medium alone (Ctrl), IgG beads (1:1 ratio), CD40 L (1 µg/mL), TNF (10 ng/mL) and LPS (1 µg/mL, 100 ng/mL, 10 ng/mL) for 24 h, in the absence (med, performed only with unstimulated cells) or presence of carrier DMSO or RN486 (Btki, 1 μM) as assessed by ELISA. Bars represent the means and SEM of seven independent experiments. *p<0.05, **p<0.01 versus cells exposed to DMSO. (C) Immunoblotting of lysates obtained from control macrophages (no virus) and macrophages transduced with adenoviruses encoding control GFP (ad-GFP) or Btk (ad-Btk-HN). (D) Analysis of IL-6 production in supernatants of control macrophages (no virus), and macrophages transduced with GFP (ad-GFP) or Btk (ad-Btk-HN) stimulated with medium (med) alone, IgG beads or CD40L as in A above. Bars represent the means and SEM of six independent experiments. *p<0.05 compared to cells transduced with GFP alone.
To obtain a broader assessment of Btk-dependent macrophage gene expression which might be relevant to RA, we stimulated macrophages (n=3 independent donors) with IgG beads, CD40L or 10% RA SF in the absence or presence of RN486 and determined expression levels of 238 genes involved in the regulation of innate immune responses, extracellular matrix metabolism, cell adhesion, inflammation and angiogenesis. In this qualitative screening, only genes which were up-regulated twofold or more in three independent experiments were considered for further analysis (see online supplementary tables S5–S7). Each stimulus generated an unique but overlapping profile of induced genes, and only four genes involved in IL-1 signalling, IL1B, IL36B, IL1RN and IL36RN, were induced by all three stimuli (figure 4A). In each of the three experiments, RN486 inhibited by more than twofold 12 of 21 genes induced by IgG, 11 of 52 genes induced by CD40 L, and 6 of 25 genes induced by 10% RA SF (figure 4B). These putative Btk-regulated genes included ones that were uniquely inhibited in response to a specific agonist, such as IL1B (IgG stimulation), CXCL9 (CD40L) and CCR3 (RA SF), as well as those inhibited in response to multiple stimuli, such as IL-1 signalling components, TNF, MMP1 and MMP10 (figure 4C).

Identification of genes regulated by IgG, CD40L and rheumatoid arthritis (RA) synovial fluid stimulation of human macrophages and their regulation by Btk. (A) Venn diagram of genes that are induced by more than twofold in three independent experiments in macrophages obtained from different donors stimulated for 4 h with IgG beads (1:1 ratio), CD40L (1 µg/mL), and 10% RA synovial fluid (SF RA, pooled from five RA patients) compared to medium alone. Genes induced by specific or groups of stimuli are listed. (B) Diagrams showing overlap in genes that are more the twofold up-regulated by indicated stimuli and more than twofold up- or down-regulated upon Btk inhibition. (C) Venn diagram of IgG, CD40L and RA SF inducible genes that were suppressed by more than twofold by RN486 (Btki, 1 μM) in three independent experiments in human macrophages compared to DMSO alone.
Btk inhibition blocks CD40-induced inflammatory mediator production in macrophages and RA synovial explants
To independently validate these findings, we assessed macrophage culture supernatants for candidate secreted proteins. Of the analytes assessed, RN486 failed to significantly influence their induction by either IgG or TNF (figure 5A). However, CD40L-induced macrophage production of TNF, MMP-1, MMP-10 (all p<0.05) and IL-10 (p<0.01) was significantly inhibited by RN486, while production of IL-1RA, IL-12p40, CXCL5, CXCL10 and MMP-7 was unaffected (figure 5A). Surprisingly, RN486 significantly reduced levels of LPS-induced MMP-1, MMP-7 and MMP-10 (all p<0.05). IL-1α, IL-1β, IFNγ, VEGF and CXCL-11 were not produced by macrophages at detectable levels in these experiments. To determine if the observed contributions of Btk to macrophage activation might also be relevant to expression of inflammatory mediators in RA synovial tissue, we cultured RA patient (n=8) synovial explants in the absence and presence of RN486. RN486 significantly decreased synovial explant production of IL-6 (p<0.01), PDGF-AA (p<0.01), CXCL-9 (p<0.05) and MMP-1 (p<0.05), but not IL-1RA, VEGF, CXCL-5, CXCL-6, CXCL-10 and MMP-10 (figure 5B), while supernatant concentrations of IL-10, TNF, IL-12p40 and MMP-7 were below the detection limits of the assay (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of RN486 on the production of inflammatory mediators by human macrophages and rheumatoid arthritis (RA) patient synovial tissue explants. (A) Analysis of indicated cytokines, soluble receptors, matrix metalloproteinases and chemokines in supernatants of macrophages stimulated with medium alone (Ctrl), IgG beads (1:1 ratio), CD40L (1 µg/mL), TNFα (10 ng/mL) and LPS (1 µg/mL) for 24 h, in the absence (med, performed for unstimulated cells only) or presence of carrier DMSO or RN486 (Btki, 1 μM) as assessed by single-plex assays. Supernatants assessed here are the same as those used for measuring IL-6 and IL-8 in figure 3. Bars represent the means and SEM of seven independent experiments. *p<0.05, **p<0.01 versus cells exposed to DMSO. (B) Analysis of indicated cytokines, soluble receptors, matrix metalloproteinases and chemokines in supernatants of RA patient (n=8) synovial tissue explants cultured for 48 h in the absence (carrier DMSO only) or presence of RN486 (1 μM) as assessed by ELISA (IL-6 and IL-8) or single-plex assays. Data for each patient were adjusted for biopsy weight and were normalised to 100 for tissue exposed to carrier only, and data expressed as % analyte concentration of RN486-treated tissue compared to control. Each circle represents data from an individual patient and bars represent the median values. *p<0.05 compared to control samples.
Discussion
Btk plays critical roles in B cell development and antigen-dependent activation of mature B lymphocytes, and also in the inflammatory activation of myeloid lineage cells.10 ,31 Accordingly, pharmacological interference with Btk signalling protects against pathology in both B cell-dependent and myeloid cell-dependent models of experimental arthritis. In murine CIA, a model critically dependent upon B cell activation and autoantibody production, prophylactic or therapeutic administration of Btk inhibitors reduces autoantibody production, synovial inflammation, and cartilage and bone destruction.20–23 ,25 Btk inhibitors similarly protect against synovial inflammation and joint destruction in murine autoantibody-induced arthritis and rat adjuvant-induced arthritis, models in which myeloid cells are primary contributors to pathology.20 ,22 ,25 Here, we demonstrate that Btk is expressed and contributes to the maintenance of inflammation in the synovium of patients with established RA.
Btk is expressed at equivalent levels in the synovium of patients with RA and PsA, and consistent with known physiological expression patterns, we observe that Btk protein is expressed in B cells, macrophages and mast cells in RA synovial tissue. Btk was also readily detected in synovial macrophages of PsA patients, but Btk expression levels did not correlate with synovial macrophage numbers in PsA, possibly due to the smaller size of this cohort. Although LPS has been reported to induce Btk expression in RA FLS in vitro when microRNA-346 induction is suppressed, we were unable to detect Btk expression in RA FLS in vivo or in vitro.32 Thus, based on cellular prevalence, our data indicate that macrophages would be the most prominent direct synovial cell population targeted by eventual application of Btk inhibitors for the treatment of RA. In other tissue compartments, such as lymph nodes and bone marrow, B cells and osteoclasts are likely to represent additional potential targets.14 ,17
To examine the potential contributions of macrophage Btk signalling to inflammation, we assessed the effect of a highly specific Btk inhibitor, RN486, on macrophage responses to agonists relevant to their activation in inflammatory arthritis. RN486 displays a more than 100-fold inhibitory selectivity for Btk over other kinases, including Tec family members, prevents pathology in murine CIA and rat adjuvant-induced arthritis, and suppresses FcγR-mediated TNF production in human monocytes.22 This latter observation might be particularly relevant to a role for Btk in the pathology of RA, as RF and ACPA immune complex activation of FcγRIIa can promote macrophage TNF production.33–35 While we observed that RN486 could significantly but partially suppress IgG-induced IL-6 production by human macrophages, only trends toward reduced TNF and MMP-7 production were observed. These results are consistent with previous observations that FcγR signalling in mature human macrophages displays a less stringent requirement for Btk involvement than in human monocytes or murine macrophages.20 ,25
Activation of toll-like receptor (TLR) expressed by synovial macrophages and FLS is thought to make important contributions to pathology in RA.36 Studies using monocytes and macrophages from XLA patients, XID mice, and treatment of healthy donor human and wild-type murine macrophages with Btk siRNA and Btk inhibitors have provided conflicting interpretations as to whether Btk is required37–39 or dispensable20 ,22 ,25 ,40 for mediating TLR2 - and TLR4-induced inflammatory cytokine production. These differences might be explained by different requirements for Btk signalling during myeloid differentiation and between human and mouse, as in the case of FcγR signalling. However, it is also noteworthy that in the above-described studies, readouts for cellular activation were largely limited to IL-6, IL-8 and TNF production. In our studies, we failed to observe an effect of RN486 on LPS-induced production of these cytokines. However, we do find that RN486 selectively and significantly inhibits macrophage production of MMP-1, MMP-7 and MMP-10 in response to LPS.
CD40, responding to stimulation by CD40L during cell–cell contact, activates many cell populations found in RA synovial tissue, including lymphocytes, macrophages, dendritic cells and FLS, and therapeutic strategies specifically targeting CD40 expressed by myeloid cells has shown to be effective in CIA.41 ,42 Potential contributions of CD40 to pathology in RA are underscored by identification of SNP variants in the CD40 gene locus, and in genes encoding signalling proteins downstream of CD40, that constitute an extended risk pathway.43 ,44 For these reasons, we were intrigued by recent data indicating that coupling of TLR signalling to Btk in macrophages occurs not at the cellular surface membrane but in endosomal compartments following TLR internalisation, and requires Btk association with major histocompatability class II proteins via CD40.39 As CD40 signalling in endothelial cells has been reported to require endosomal internalisation, and CD40 regulates Btk activity in B cells, we considered whether Btk inhibition may also influence CD40-dependent activation of macrophages.45 ,46 Indeed, we find that RN486 significantly reduces CD40L-induced IL-6, IL-8, MMP-1 and MMP-10 production, a profile which overlaps with, but is distinct from, the effect of RN486 on LPS-induced gene expression in macrophages. Treatment of RA synovial explants with RN486 reduced spontaneous production of gene products associated with macrophage Btk involvement in response to CD40L (IL-6, IL-8, MMP-1, PDGF-B) and unidentified components of RA SF (CXCL-9), possibly indicating that these are predominant pathways driving macrophage Btk activation in RA synovial tissue. The abundant expression of Btk in RA and PsA synovial macrophages, the pro-inflammatory involvement of Btk in macrophage responses to multiple stimuli, and the strong association between synovial macrophages and disease activity47 as well as the consistent relationship between reductions in macrophages and clinical improvement after effective therapy in RA48 suggest that continued development of Btk inhibitors may be of potential therapeutic benefit in the treatment of RA, as well as other forms of inflammatory arthritis in which macrophage activation contributes to pathology.
Acknowledgments
We would like to thank C. van der Horst (Academic Medical Center, University of Amsterdam) for help with digital image analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
- Data supplement 5 - Online table 3
- Data supplement 6 - Online table 4
- Data supplement 7 - Online table 5
- Data supplement 8 - Online table 6
- Data supplement 9 - Online table 7
Footnotes
Handling editor Tore K Kvien
Contributors LMH contributed to research design, performed experiments, analysed data and contributed to writing the paper; IEvE performed experiments and analysed data; MWT obtained, recorded, analysed and interpreted all clinical records and materials, JSF, MS, JW, SN, JD, PPT and KAR designed the research, analysed and interpreted data, and contributed to writing the manuscript.
Funding This research was supported by an open research programme grant from Hoffman-La Roche, Inc. to KA Reedquist.
Competing interests This research was supported by an open research programme grant by Hoffman-La Roche, Inc. to KAR. JSF, MS, JW, SN and JD are employees of and shareholders in Hoffmann-LaRoche, Inc.
Ethics approval The Medical Ethics Committee, Academic Medical Center, University of Amsterdam approved this study.
Provenance and peer review Not commissioned; externally peer reviewed.