Article Text

Abstract
Objective Altered microbiota composition or dysbiosis is suspected to be implicated in the pathogenesis of chronic inflammatory diseases, such as spondyloarthritis (SpA) and rheumatoid arthritis (RA).
Methods 16S ribosomal RNA gene sequencing was performed on faecal DNA isolated from stool samples in two consecutive cross-sectional cohorts, each comprising three groups of adult volunteers: SpA, RA and healthy controls (HCs). In the second study, HCs comprised a majority of aged-matched siblings of patients with known HLA-B27 status. Alpha and beta diversities were assessed using QIIME, and comparisons were performed using linear discriminant analysis effect size to examine differences between groups.
Results In both cohorts, dysbiosis was evidenced in SpA and RA, as compared with HCs, and was disease specific. A restriction of microbiota biodiversity was detected in both disease groups. The most striking change was a twofold to threefold increased abundance of Ruminococcus gnavus in SpA, as compared with both RA and HCs that was significant in both studies and positively correlated with disease activity in patients having a history of inflammatory bowel disease (IBD). Among HCs, significant difference in microbiota composition were also detected between HLA-B27+ and HLA-B27 negative siblings, suggesting that genetic background may influence gut microbiota composition.
Conclusion Our results suggest that distinctive dysbiosis characterise both SpA and RA and evidence a reproducible increase in R. gnavus that appears specific for SpA and a marker of disease activity. This observation is consistent with the known proinflammatory role of this bacteria and its association with IBD. It may provide an explanation for the link that exists between SpA and IBD.
- spondyloarthritis
- rheumatoid arthritis
- ankylosing spondylitis
- infection
- inflammation
Statistics from Altmetric.com
Introduction
Spondyloarthritis (SpA) is a multifactorial heterogeneous disorder, which is thought to result from complex interactions between a particular genetic background and environmental factors. The clinical spectrum of SpA is diverse, comprising both axial and peripheral joint inflammation and extra-articular manifestations, including psoriasis, uveitis and inflammatory bowel disease (IBD).1 Heredity is high in SpA, and several genetic polymorphisms have been shown to influence the risk of this disorder. The most important one is the MHC class I allele HLA-B27.2 Several other polymorphisms have been discovered in the recent years, through genome-wide association studies, pointing towards putative pathophysiological pathways. Remarkably, a large subset of the responsible genes code for proteins involved in immune response, and particularly in the interleukin (IL)-23/Th17 pathway of T cell differentiation, which is primarily implicated in response against extracellular pathogens, including bacteria and yeasts, and/or in microbial sensing.3 Moreover, cross-disease genetic association study established that a substantial fraction of SpA heritability is due to genetic factors that also predispose to IBD.4
In contrast to genetic factors, the role of external influences on disease development and/or evolution has been far less studied in SpA. This is particularly the case regarding the putative influence of the abundant gut microbiota content. Disequilibrium in such complex realm of microbes that closely interact with the gut mucosal immune system, a situation referred to as dysbiosis, has been associated with several chronic inflammatory disorders and in particular with IBD, including Crohn’s disease and ulcerative colitis.5
The role of microbiota in animal models of arthritis has been established for decades. It is notably the case of the HLA-B27/human β2-microglobulin transgenic rat model of SpA, in which gut dysbiosis was found and breeding under germ-free conditions prevented the development of both arthritis and the accompanying IBD phenotype mimicking ulcerative colitis.6 More recently, several pioneering microbiota high-throughput sequencing studies conducted in human arthritides also reported gut dysbiosis either in stool samples from patients affected by rheumatoid arthritis (RA), psoriatic arthritis (PsA) or juvenile enthesitis-related arthritis, a paediatric form of SpA, or in ileal biopsies from adult patients with SpA.6–8 Despite showing promising results, the studies that concerned PsA and SpA were each of relatively small size (9–27 patients compared with 9–17 healthy controls (HCs)), and the differences between patients and controls were therefore of limited statistical significance with regard to the number of microbial species examined.9
In an attempt to further interrogate if specific gut dysbiosis may associate with SpA, we performed metagenomic analysis, comparing stool samples between adult SpA and control groups, that included both healthy individuals and patients with RA. We used a two-stage design that allowed us to test for the reproducibility of our results. Furthermore, the second phase of the study included a large number of healthy sibling of patients with SpA as controls, allowing us to control in as much as possible for genetic background variability and to interrogate HLA-B27 effect.
Patients and methods
Ethics statement
The study was conducted according to the Declaration of Helsinki and French legislation. Before study inclusion, each participant gave written informed consent for research use and publication of their data.
Population recruitment and study design
The study population was recruited in two consecutive phases. In the first discovery phase, patients having established SpA or RA and consulting in a tertiary-care centre, that is, the rheumatology clinic of Ambroise Paré Hospital, were included, as well as HCs belonging mostly to the hospital staff. In the second confirmatory phase, patients identified as having SpA and belonging to French families collected by Groupe Français d’Etude Génétique des Spondyloarthrites for genetic purposes as previously described,10 were included as well as aged-matched healthy siblings. Recently diagnosed patients with RA having not yet received corticosteroid, disease-modifying antirheumatic drug (DMARD) or biotherapy were recruited from Ambroise Paré Hospital outpatient clinic.
Altogether, stool samples were collected from 199 volunteers consisting of 96 SpA patients, all fulfilling the Assessment of SpondyloArthritis International Society (ASAS) classification criteria,11 32 independent patients with RA fulfilling the American College of Rheumatology/European League Against Rheumatism classification criteria12 and 71 HCs, including 28 genetically independent controls and 43 who were recruited as siblings of patients with SpA enrolled in the study (22 HLA-B27+ and 21 HLA-B27− siblings).
All participants were required not to have received antibiotics during the preceding month, nor to have undergone gut preparation for colonoscopy during the preceding 6 months. Individuals affected with medical condition other than the diseases under investigation thought to potentially affect results of the study were excluded. This concerned notably other osteoarticular inflammatory disorders and/or autoimmune disorders, obesity, diabetes, cancer and any chronic organ failure. Demographic data, body mass index, clinicobiological data relevant for disease classification or disease activity evaluation (ie, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) as for SpA and Disease Activity Score 28 (DAS28)-erythrocyte sedimentation rate as for RA, respectively) and current treatments were collected. Active disease was defined as a BASDAI ≥3/10, a cut-off corresponding to the lower limit of the ‘patient acceptable symptomatic state’ 95% CI, as proposed in an earlier study13 or a DAS28 ≥3.2/10, as for SpA and RA, respectively.
Fifteen individuals were removed from analysis because of an insufficient yield of bacterial sequences (see below). Therefore, detailed characteristics of the study population are shown for the 184 individuals that were included in the final analysis (table 1).
Characteristics of patients and controls included in the final analysis
Faecal DNA isolation
Fresh stool samples were either conserved in a close bucket containing Anaerocult A system (Merck, Darmstadt, Germany) for a maximum of 4 hours before being processed and stored frozen or immediately frozen and kept at −80°C before being further processed.
Faecal DNA was extracted from the weighted stool samples as previously described.14 After the final precipitation, DNA was resuspended in 150 mL of Tris-EDTA (TE) buffer, and stored at −20°C for further analysis.
16S ribosomal RNA (16S rRNA) gene sequencing
Microbial diversity was determined for each sample by targeting a portion of the ribosomal genes. A 16S rRNA gene fragment comprising the V3 and V4 hypervariable regions (16S (sense) 5′-TACGGRAGGCAGCAG-3′ and (antisense) 5′-CTACCNGGGTATCTAAT-3′) was amplified using an optimised and standardised 16S-amplicon-library preparation protocol that gives the lowest error rates. Barcode sequences (GsFLX key) TCAG and MIDGsFLX (10 nucleotides) were attached between the 454 GsFLX adaptator sequence and the forward primer V3F. The GsFLX key and the 454 GsFLX adaptator were attached to the reverse primer. The concentration and quality of the PCR products were assessed with Picogreen in order to obtain equal amounts of each of the samples (108 molecules/mL), and then 16S rRNA gene amplicons were sequenced on a Roche GS FLX 454 sequencer (Genoscreen, Lille, France) and processed with standard protocol from manufacturer (http://genoscreen.fr/).
16S rRNA gene sequence analysis
The analysis was performed as described previously.15 The sequences were demultiplexed and quality filtered using the ‘quantitative insights into microbial ecology’ (QIIME, V.1.8.0) software package.16 The sequences were assigned to operational taxonomic units (OTUs) using the UCLUST algorithm17 with a 97% threshold of pairwise identity and classified taxonomically using the Greengenes reference database.18 Rarefaction was performed (2028–50 076 sequences per sample; 15 samples with less than 2000 sequences were excluded from analysis) and used to compare the abundances of OTUs across samples.
Statistical plan
Two series of samples, each including patients with SpA and RA and HCs were recruited successively, as described above, and processed in two different runs of sequencing that were analysed separately.
Principal component analyses of the Bray Curtis distance with each sample coloured according to the disease phenotype were built and used to assess the variation between experimental groups (beta diversity). The number of observed species as well as the Shannon, Simpson and Chao1 diversity indexes were calculated using rarefied data (depth=2000 sequences/sample) and used to characterise species diversity in a community. Differential analysis was performed using the linear discriminant analysis effect size (LEfSe) pipeline.19
MaAsLin, a multivariate statistical framework, was used to find associations between treatment and microbial community abundance.20 The following parameters were taken into account in the analysis: age, gender, disease status (SPA, RA and HCs), definite radiographic sacroiliitis (modified New York criteria), B27 genotype, disease activity, association with IBD and treatments (including biotherapy, non-steroidal anti-inflammatory drug (NSAID), corticosteroid, methotrexate, sulfasalazin and proton pump inhibitor). Associations were considered significant for p value <0.05 and q value <0.1.
Mann-Whitney test was used to compare Ruminococcus gnavus abundance between groups, and non-parametric Spearman’s test was used to test the correlation between R. gnavus relative abundance and BASDAI. A two-way analysis of variance was used to compare R. gnavus abundance between SpA patients with and those without a history of IBD, stratifying for disease activity.
Results
Discovery study
In the discovery study, faecal microbiota composition was analysed in 49 SpA, 17 RA and 18 HC. Beta diversity analysis showed that the microbiota composition was significantly different between the three groups (Bray Curtis index, Anosim 9999 permutations, p=0.005; see online supplementary figure 1A). Both SpA and RA differed from HCs (p=0.02 and p=0.003, respectively; figure 1A, B) as well as SpA from RA (p=0.03; see online supplementary figure 1B). The alpha diversity assessed by the number of observed species was significantly decreased in both SpA and RA, as compared with HCs (p<0.001; figure 1C). There was a trend towards more reduced diversity in RA than SpA (p=0.08; figure 1C). The reduced alpha diversity was not correlated with disease activity, as shown for SpA (see online supplementary figure 2).

Bacterial composition of the gut microbiota was significantly different between disease groups and HCs in the discovery cohort. (A and B) Beta diversity according to Bray Curtis index was significantly different between SpA and HCs (A) and between RA and HCs (B). (C) Alpha diversity as assessed by the observed number of bacterial species was significantly reduced both in SpA and RA, as compared with HC. (D and E) Variations in the bacteria phyla (D) and families (E) profiles were apparent between patient groups and HCs. HCs, healthy controls; PC, principal component; RA, rheumatoid arthritis; SpA, spondyloarthritis.

Bacterial taxa that were differentially represented in the three studied groups, that is, SpA, RA and HCs, in the discovery cohort, with statistical level of significance according to linear discriminant analysis (LDA score >2). The histogram displays all taxa that were increased in each group, as compared with both others, and the corresponding level of significance (LDA score). Taxa were identified at the order, family, gender or species level and colour-coded according to their phylum, and the legend is shown on the right-hand side. HCs, healthy controls; RA, rheumatoid arthritis; SpA, spondyloarthritis.
In all groups, the profile of gut microbiota appeared dominated by Firmicutes and Bacteroidetes at the phylum level and by Lachnospiraceae, Ruminococcaceae and Bacteroidaceae families, consistent with the usual composition of the human gut microbiome (figure 1D). However, some variations in the distribution were apparent between groups, including more Lachnospiraceae in SpA, less Prevotellaceae and Paraprevotellaceae in SpA and RA than in HCs, less Bifidobacteriaceae in RA and more Proteobacteria, including Enterobacteriaceae, in RA than in both other groups.
Discriminant analysis using LEfSe identified significant taxa variations distinguishing each sample group from both others (figure 2). Patients with SpA had an increase in Firmicutes belonging to the Lachnospiraceae family, including Ruminococcus, Dorea, Coprococcus and Blautia genera and in Actinobacteria from the Coriobacteriaceae family. Increased species included R. gnavus, Blautia pruducta and Bifidobacterium longum. At the OTU level, we observed significant differences between SpA and controls, including increased amount of several Blautia, Ruminococcus and Coprococcus OTU and decreased amount of several Roseburia faecis OTU in SpA (see online supplementary figure 3).

(A) Bacterial composition of the gut microbiota was significantly different between HLA-B27+ SpA cases and their matched healthy siblings in the replication cohort, according to Bray Curtis index (beta diversity), (B) but not between matched HLA-B27+ and HLA-B27− healthy siblings. PC, principal component.
In contrast, patients with RA had an increase in Proteobacteria, including Klebsiella genus, Desulfovibrionaceae and Succinivibrionaceae families, and in the Tenericutes and Synergistetes phyla (figure 2). Finally, HCs were characterised by significantly more Actinobacteria of the Bifidobacteriaceae family, Proteobacteria of the Oxalobacteraceae family, R. faecis and an unidentified Prevotella species (figure 2).
Multivariate statistical analysis using MaAsLin identified some treatment effects, mostly related to sulfasalazine intake, that were distinct from disease status-related variations (see online supplementary table 1).
Confirmatory study
This foregoing discovery study revealed striking differences between the three studied groups, indicative of distinct dysbiosis in both SpA and RA. This prompted us to undertake a replication study in which we included a majority of siblings of patients with SpA as HCs, allowing somehow to control for the genetic background and in particular for the HLA-B27 status shared between patients and half of their siblings. Besides disease status, confounding factors, such as immunomodulatory treatments, could have impacted results of the discovery study, especially in patients with RA, a majority of whom were receiving such treatments. This is why we only included this time patients with early RA having never been treated with corticosteroid, DMARD or biotherapy.
The overall microbiota profile was broadly comparable with that seen in the discovery cohort, although this time, the SpA group appeared more similar to the healthy controls (online supplementary figure 4). The RA had less Firmicutes of the Ruminococcaceae family and again less Bacteroidaceae but more Proteobacteria of the Enterobacteriaceae family than both other groups (see online supplementary figure 4). In the family-based study, comparison of a group of 29 patients with SpA (all B27+) with their 41 matched siblings showed again that, based on beta diversity index, the composition was significantly different between SpA and HCs (Bray Curtis index, Anosim 9999 permutations, p=0.02; figure 3A), whereas it was not different between the B27+ and B27− healthy siblings (p=0.3; figure 3B). Using LEfSe, the most significantly increased taxa in the SpA group was R. gnavus, which had already been identified in the discovery cohort (figure 4A). Besides, a majority of the differentially expressed taxa were decreased in SpA. Among healthy siblings of SpA, the most significant differences between HLA-B27+ and B27− controls consisted of an increase in Microcaccaceae and Rothia mucilaginosa and a decrease in Bifidobacterium genus and Odoribacter species in B27+ siblings (figure 4B). At the OTU level, increased amount of several Blautia, Ruminococcus and of Eggertthella lenta and decreased amount of several Bifidobacterium characterised HLA-B27+ siblings (see online supplementary figure 5).

Bacterial taxa that were differentially represented between groups in the replication cohort, with statistical level of significance according to linear discriminant analysis (LDA score >2). (A) Histogram of the taxa that were significantly different between HLA-B27+ SpA patients (n=29) and matched healthy siblings (n=41). (B) Histogram of the taxa that were significantly different between HLA-B27+ healthy siblings of patients (n=22) and matched HLA-B27− healthy siblings (n=21). (C) Histogram of the taxa that were significantly over-represented in the whole SpA, RA and HC groups, as compared with both other groups. Taxa were identified at the order, family, gender or species level and colour-coded according to their phylum (same colour code as in figure 2).
![[SP1.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1614/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
![[SP2.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1614/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
![[SP3.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1614/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
![[SP4.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1614/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
![[SP5.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1614/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
{kind=link}
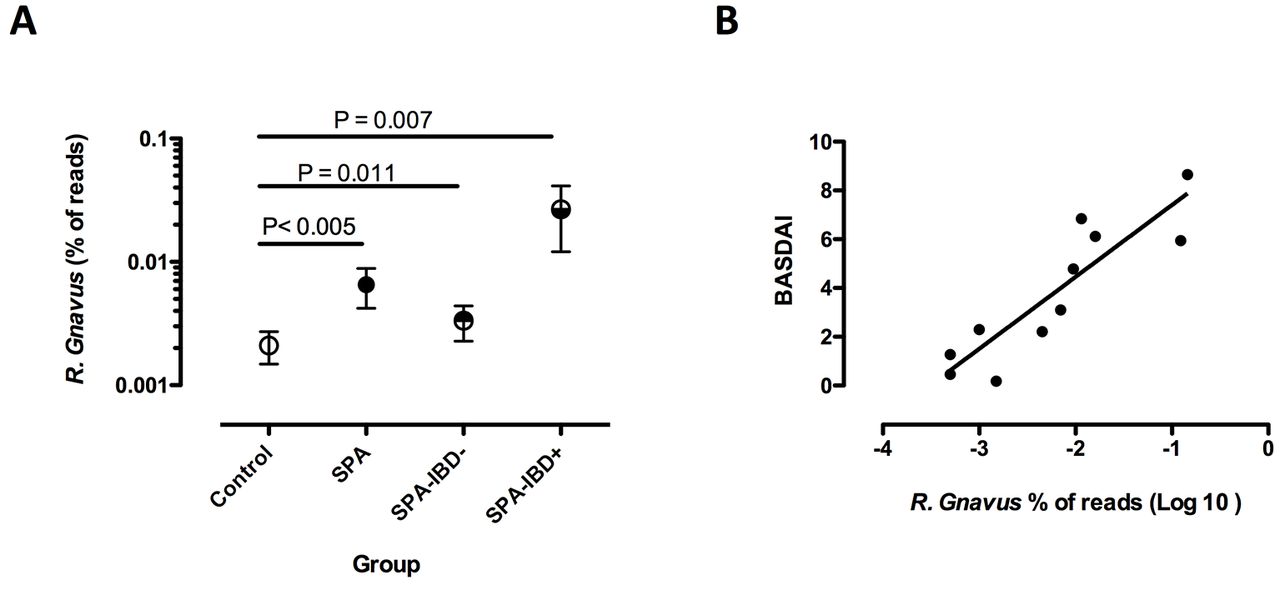
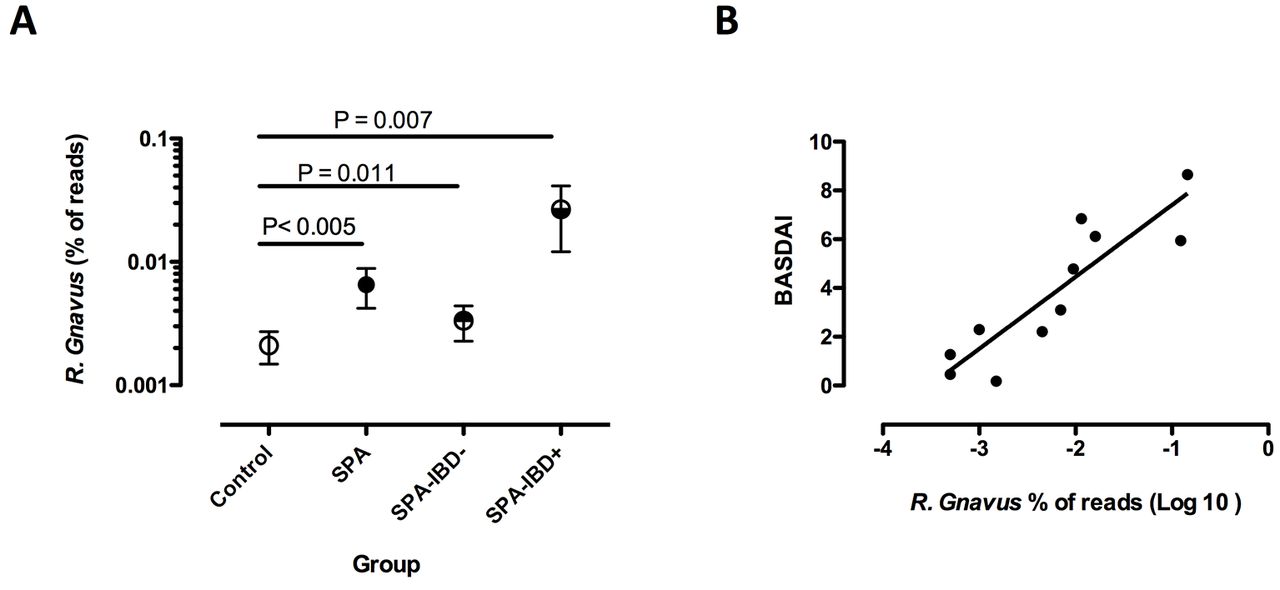
Ruminococcus gnavus was significantly over-represented in the gut microbiota of SpA patients, with (IBD+) or without (IBD−) a positive history of IBD (A). R. gnavus abundance was significantly correlated with the BASDAI in SpA patients with a positive history of IBD (B). Symbols in (A) represent the proportion of R. gnavus reads among all bacteria in faecal samples, expressed as a mean percentage and SE of the mean of all reads in each group. BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; IBD, inflammatory bowel disease; SpA, spondyloarthritis.
In the next step, we analysed together all the samples collected in the replication study, consisting of SpA (n=38), family and unrelated HCs (n=51) and RA of recent onset (n=11). Again, these three groups were distinguishable from each other based on LEfSe and, remarkably, the unique taxon that differentiated SpA from both RA and HCs was R. gnavus (figure 4C). This time, RA was characterised by an increase in unidentified Lactobacillus species, Corynebacterium variabile, Staphylococcus aureus, Facklamia and Paraprevotallaceae. HCs were characterised by a greater abundance of an unidentified Anaerostipes species.
R. gnavus abundance correlates with BASDAI in patients with SpA having IBD history
We further analysed the influence of clinical parameters associated with SpA status on the increased abundance of R. gnavus in the combined studies. Increased R. gnavus was significant only in active SpA, predefined by a BASDAI ≥3/10 (p=0.034). R. gnavus was increased in SpA patients without IBD (n=74; p=0.012) as well as in those having a history of IBD (n=12; p=0.007), as compared with HCs (figure 5A). However, stratifying on disease activity, the increase in R. gnavus appeared of greater magnitude in patients with IBD history than in those without (mean percentage of R. gnavus reads: 0.0267 vs 0.0033, respectively; p=0.001). Moreover, there was a striking correlation between R. gnavus abundance and BASDAI in the subgroup of patients having IBD history (p<0.005, r=0.77; figure 5B) but not in the others (data not shown). Interestingly, this was not related to IBD activity, since 8 out of the 10 patients with SpA having a history of IBD, for which the information was available, were in remission of IBD at the time of faecal sampling and the two who had active IBD had low BASDAI activity (1.3 and 2.2, respectively). Noteworthy, there was no correlation between abundance of R. gnavus and NSAID intake.
Discussion
Altered gut microbiota composition or intestinal dysbiosis is a possible actor in chronic inflammation, even in distant sites, such as the joint.21 However, until now, only few studies have addressed this question.6 Here we report a two-stage study, including a discovery and a replication steps that compared for the first time gut microbiota composition between the two most common causes of chronic inflammatory disorders of the joint, that is, SpA and RA, and HCs.
We first identified dysbiosis in both arthritic disorders but with striking differences between them, indicating that it was not a mere consequence of the inflammatory state, but rather bore disease specificity. One of the main changes that was observed in both diseases was a reduced microbial diversity, a consistent finding in microbiota studies of chronic inflammatory disorders20 that was also reported in faecal microbiota of patients with PsA.22
However, the most striking finding of our study was a significant enrichment in R. gnavus, which specifically distinguished SpA from other groups (RA and HCs) in both cohorts. In the replication cohort, R. gnavus was significantly enriched in patients with SpA, even as compared with matched healthy siblings. The latter group was included, since it was expected to have microbiota profile more similar to patients than unrelated HCs, except for disease-related differences, due to a greater share of genetic and environmental factors.23 24
To our best knowledge, this is the first metagenomic study to report a significant and reproducible enrichment in a gut-resident bacterial species in SpA. R. gnavus is a strict anaerobic Gram-positive non-spore-forming cocci that belongs to the Lachnospiraceae family and is a frequent commensal of the gut.25 It belongs to a limited consortium of bacteria that display mucolytic activity on mucin-2, the principal constituent of intestinal mucus, due to their glycosidases activity.26 Such property may facilitate their association with the mucosa and close location to the epithelial border. R. gnavus also expresses β-glucuronidase activity that can generate toxic metabolites in the colon that might provoke local inflammation.27 Additionally, it exhibits high bile acids 7α-dehydroxylating activity, leading to the production of secondary bile acids, that is, deoxycholic and lithocholic acids, and 3α-hydroxysteroid dehydrogenase required to convert bile acids into isobile acids, that is, isocholic, isodeoxycholic and isolithocholic acids, which may alter microbiota composition by detoxifying deoxycholic acid.28
Its increased abundance in faeces and mucosa has been associated with Crohn’s disease, with the recurrence of Crohn’s disease after ileocolonic resection and with the risk of ulcerative colitis on anastomosis, that is, pouchitis.29–33 In IBD, it was preferentially associated with the gut mucosa, which confers to this mucolytic bacteria a possible role in the triggering or maintenance of inflammation.31 34 This is particularly relevant to our study, since it was mainly over-represented in patients with active SpA. Remarkably, it was most abundant in the subgroup of patients having a history of IBD and its abundance positively correlated with SpA activity in those patients, even though IBD was inactive at the time of sampling in most of them. This suggests that the abundance of R. gnavus could be more or less directly involved in osteoarticular inflammation and may provide some insight into the pathogenic link that exists between SpA and IBD.
A direct role for gut microbiota in the triggering of SpA is strongly supported by animal models. In HLA-B27 transgenic rats, the spontaneous development of both arthritis and IBD was prevented by raising animals in germ-free conditions, and those manifestations were triggered by recolonising the germ-free rats with a limited consortium of bacterias and even with single bacterial species.35 In those experiments, Bacteroides vulgatus, which has been shown as increased in the HLA-B27 transgenic rat gut was sufficient to trigger both IBD and arthritis.35 36 B. vulgatus was not increased in our study, but it is interesting to underline that this bacterial species is one of a few having mucolytic activity and that it could degrade porcin but not human mucin, showing that a bacterial species may bear distinct biological consequences, according to the colonised animal species.34 Remarkably, Akkermansia muciniphila, which is another mucolytic bacterial species, was also reported as increased in the gut of HLA-B27 transgenic rat in correlation with local expression of proinflammatory cytokines, including interferon-γ, IL-17A, and IL-23, and with the development of arthritis,37 as well as in a subgroup of children with enthesitis-related arthritis, an early form of SpA.38 Altogether, those observations highlight that distinct bacterial species that have been shown as associated with SpA in different settings share mucolytic activity. Such property could be an essential trigger of disease pathogenesis by facilitating access of the gut epithelium to other commensal bacteria and their invasion inside the mucosa that may contribute to distant joint inflammation.39
In our study, we examined the bacterial composition of stool samples, whereas two previous studies performed in SpA were focused on the mucosal-associated microbiota.7 40 The first of those studies compared the terminal ileum-associated microbial community between 10 ankylosing spondylitis patients and nine HCs. Striking differences at the phylum and gender bacterial levels were reported between both groups, some of which were consistent with our findings, such as an enrichment of the Firmicutes phylum and more specifically of the Lachnospiracae family, including Coprococcus species and Ruminococcaceae, in SpA. Also an increase in the secondary bile acid biosynthesis pathway was consistent with an increase in Ruminococcaceae. However, the microbial diversity was increased in SpA, as opposed to our results, a finding that is difficult to explain as inflammation have been reported in human and animals to be associated with a decreased biodiversity. The second study included ileal and colonic biopsies from 27 SpA, half of them displaying microscopic bowel inflammation at the time of sampling, and 15 HCs. Overall, there was no significant difference in bacterial composition between both groups but a trend towards an increased richness in the inflamed samples. Besides, there was also an increased abundance of the Dialister genus in the inflamed biopsies, which correlated with disease activity index.7
Studying mucosal specimens may give access to the bacterial community in close interaction with the intestinal tissue and its immune system, the most likely to be directly involved in disease pathogenesis. However, it requires an invasive procedure and a simultaneous evaluation of the histological aspect of the mucosal biopsies, whereas studying stools allowed us to readily collect large numbers of samples following reproducible protocol. Moreover, we included patients having established disease and, in the context IBD, it was shown that if dysbiosis was more readily detected in the mucosal-associated bacterial community in early disease, it was spreading to the lumen content in advanced disorder, allowing to catch similar variations on stools than mucosal biopsy samples in established disease.41 42
Environmental factors known as affecting microbiota, such as breastfeeding, have been shown to influence SpA susceptibility.39 Given the high heredity in SpA, genetic factors may also contribute to the development of dysbiosis. In support of such hypothesis, the HLA-B27+ healthy siblings of patients with SpA exhibited a microbiota composition distinct from their HLA-B27− healthy siblings, with increased R. mucilaginosa and E. lenta, two bacterial species that have been associated with IBD42 43 and low levels of Bifidobacterium and Odoribacter, similar to what has been reported in patients with ileal Crohn’s disease and ulcerative colitis.20 42
We evidenced dysbiosis in RA that was distinct from that seen in SpA. RA-associated dysbiosis differed also between both cohorts, a finding that could be attributed to the characteristics of patients with RA recruited in those cohorts. Hence, in the discovery study, patients with RA had long-standing disease and a majority of them received immunosuppressive treatment, including corticosteroids, disease-modifying antirheumatic drugs and biotherapies. Besides, their disease was moderately active in average. This population had increased proportions of aerotolerant Proteobacteria, as frequently reported in other inflammatory disorders, such as IBD, and Synergistetes, which have been associated with periodontal inflammation.44 In contrast, patients with RA included in the second cohort had short disease duration, no immunosuppressive treatment and more active disease. Their microbiota was enriched in a majority of aerotolerant Gram-positive bacteria belonging to the Firmicutes phylum, including the Lactobacillus genus and cocci of the Facklamia and Staphyococcus genera, and to the Actinobacteria phylum, such as C. variabile. Interestingly, Gram-positive bacteria, including Lactobacillus sp, have been shown as pathogenic in animal models of RA and increased in the gut microbiota in correlation with disease activity in such patients.8 9 The Paraprevotellacea family was also enriched in our study. Interestingly, Prevotella copri, a member of that family, was previously reported as increased in recent onset RA but not in long-standing RA.45
In conclusion, specific dysbiosis allowed us to distinguish SpA from HCs and from RA, whatever the stage of the latter disease. Moreover, we found evidence tha R. gnavus could be involved in SpA activity and thereby contribute to the link between SpA and IBD. Finally, HLA-B27 may contribute to gut dysbiosis and thereby to SpA predisposition.
Acknowledgments
We acknowledge the kind contribution of Mrs Nabila Yasmine Domingo-Saidji from the Clinical Research Unit of Paris Ile-de-France Ouest (URCPO), who supervised the administrative paperwork and helped us to manage ethical issues related to the study.
References
Footnotes
Contributors MB contributed to the conception and design of the work, acquisition, analysis and interpretation of data, drafting and revising of the work and final approval of the version submitted. JT contributed to the design of the work, analysis and interpretation of data, drafting of the work and final approval of the version submitted. AL contributed to the design of the work, acquisition of data and final approval of the version submitted. RS-N contributed to the design of the work, acquisition of data and final approval of the version submitted. PL contributed to the conception and design of the work, interpretation of data and final approval of the version submitted. GC contributed to the conception and design of the work, interpretation of data and final approval of the version submitted. J-PF contributed to the conception and design of the work, acquisition, analysis and interpretation of data, drafting and revising of the work and final approval of the version submitted. HS contributed to the design of the work, analysis and interpretation of data, drafting and revising of the work and final approval of the version submitted.
Competing interests None declared.
Ethics approval Comités de Protection des Personnes Paris Ile de France XI.
Provenance and peer review Not commissioned; externally peer reviewed.