Article Text

Abstract
Objective Fibrin deposits are characteristic of the synovial tissues in rheumatoid arthritis (RA). Once citrullinated, fibrin becomes an autoantigen and is thought to contribute in this way to perpetuate the disease. Our study aimed to analyse the responses of RA synovial fibroblasts (RASF) to native and citrullinated fibrin.
Methods The transcriptome induced by fibrin in RASF was approached with whole-genome-based gene expression arrays. The upregulation of selected pro-inflammatory genes by fibrin was confirmed in additional primary cell cultures using quantitative PCR and ELISA. Citrullination reactions were carried out with recombinant human peptidylarginine deiminases (PAD) 2 and 4.
Results In the whole-genome array native fibrin was found to modulate the gene expression profile of RASF, particularly upregulating mRNA levels of several pro-inflammatory cytokines. The induction of interleukin (IL)-6 and IL-8 by fibrin was confirmed in additional samples at both the mRNA and the protein level. Blocking and knockdown experiments showed the participation of toll-like receptor (TLR)4 in the induction of both cytokines. As compared with the native macromolecule, PAD2-citrullinated fibrin induced significantly higher expression of the pro-inflammatory cytokines in these cells.
Conclusions Our results suggest that fibrin mediates inflammatory responses in RASF via a TLR4 pathway. In this way, fibrin and particularly its citrullinated form may contribute to sustain the cytokine burst in RA.
- Rheumatoid Arthritis
- Fibroblasts
- Chemokines
- Ant-CCP
Statistics from Altmetric.com
Introduction
The accumulation of fibrin in joints is characteristic of rheumatoid arthritis (RA). Intra-articular fibrin can be regarded as a marker of severe disease because it is found at invasive sites,1 and has been shown to correlate with macrophage infiltration.2 Additionally, its presence at early stages of the disease suggests its role in the pathogenesis of RA.3 ,4 We have previously proposed that fibrin deposits could account for a persistent activation of surrounding RA synovial fibroblasts (RASF), conferring them with aggressive features.5 ,6
Fibrinogen, the soluble precursor of fibrin, promotes adhesiveness and migration of leucocytes through the binding of α/β integrins.7–9 The activation is likely mediated by the induction of chemokines and interleukin (IL)-6.10 In macrophages, CD14 and of toll-like receptor (TLR)4 have been shown to participate in fibrinogen-induced responses.11 ,12 TLR are detectors of pathogen-associated molecular patterns, and trigger the production of cytokines.13 TLR4 is targeted by bacterial lipopolysaccharide (LPS), but also binds endogenous products, or ‘damage’-associated molecular patterns (DAMPs), such as extracellular matrix fragments.14 Both TLR2 and TLR4 are overexpressed in RA, and it has been suggested that pathogen-associated molecular patterns and DAMPs contribute to perpetuate joint inflammation through their engagement.15 Additionally, TLR4 suppression attenuates experimental arthritis.16 ,17
Fibrin is a major autoantigen of anticitrullinated peptide antibodies (ACPA) in RA.18 ,19 Citrullination consists of the substitution of arginine residues of proteins for citrulline by peptidylarginine deiminases (PAD). Citrullination of matrix proteins can be found in inflamed joints and is not restricted to RA.20 In contrast, the development of ACPA, and also of antibodies to citrullinated fibrin (ACF), is highly specific of RA and identifies a distinct subset of patients.21 ACF are found in approximately two-thirds of the patients.22 ,23 In early arthritis subpopulations, both ACPA and ACF herald the progression to established RA, and predict a more severe course of the disease.22 ,24 ,25
Along with its contribution to the autoimmune response, citrullination could also enhance fibrin-dependent cell activation.12 ,26
We describe here the pro-inflammatory activity of fibrin in RASF via TLR4, and the stronger response elicited by its citrullinated form. Our results provide a connection between the intra-articular accumulation of fibrin, its modification by PAD and the innate inflammatory response of RA.
Materials and methods
A complete description of the experimental methods is given in the online supplementary text.
Experimental design
These studies were conducted in primary RASF cultures. Two methods of cell stimulation were employed (see online supplementary file S1, online supplementary figure S1). Activation of RASF in response to native fibrin was approached with in situ fibrin polymerisation, as described previously.27 In experiments comparing the effects of citrullinated and native fibrin, the citrullination reaction was carried out prior to cell stimulation, with PAD2 or PAD4, and cells were seeded onto the citrullinated or sham-treated fibrin clots. The efficiency of fibrin citrullination was assessed with immunoblot (see online supplementary file S1). Using these citrullination conditions, viability was studied with a tetrazolium-based colorimetric system (MTS, Sigma Aldrich, Buch, Switzerland). Controls were carried out to exclude LPS contamination of the reagents (see online supplementary file S1).
Affymetrix gene chip analysis
Total RNA was isolated from RASF (106 cells) using the MiniRNeasy kit (Qiagen, Basel, Switzerland). After reverse transcription, cDNA was used as template for hybridisation in a whole-genome Affymetrix GeneChip plate to identify genes differentially regulated by fibrin. A twofold bidirectional cut-off in gene expression levels was established to compare conditions.
Quantitative PCR
Two-step quantitative PCR studies were done with an ABI Prism 7500 Sequence Detector (Applied Biosystems, Rotkreuz, Switzerland). The relative expression was calculated by the ΔCt method, using 18S as housekeeping gene. Primer pairs are shown in online supplementary file S1.
Measurement of cytokine release
Fibrin homogenates and cell culture supernatants were prepared as detailed in online supplementary file S1. Levels of IL-8 and IL-6 were measured with commercial ELISA (BD Biosciences, Basel, Switzerland).
Blocking experiments
RASF were pretreated for 90 min with 10 μg/ml TLR4 blocking IgG2 (HTA125, Abcam, Cambridge, UK) or 10 μg/ml mouse IgG2 (BioLegend, San Diego, CA) before incubation with fibrin.
siRNA knockdown experiments
Knockdown experiments were conducted as detailed in online supplementary file S1, with small interference RNA (siRNA) targeting TLR2, TLR4, intercellular adhesion molecule (ICAM)-1, and with scrambled siRNA. The silencing efficiency on gene transcription is shown in online supplementary figure S2.
Cell immunofluorescence
Staining was performed as detailed in online supplementary file S1 and visualised with a confocal Leica SP5 microscope.
Electrophoretic mobility-shift assay
Nuclear extracts were mixed with a transcription factor (NF)κB specific consensus sequence, subjected to PAGE, and transferred to a nylon membrane (see online supplementary file S1). The presence of nuclear factor κ B (NFκB) components was confirmed by a characteristic retardation band pattern of the complexes.
Statistics
Data are expressed as average (±SE). Wilcoxon's test for paired samples was used to identify differences between conditions. p Values <0.05 were considered significant.
Results
Fibrin modulates gene expression pattern in RASF
With whole-genome gene expression arrays, we identified more than 80 overexpressed genes in RASF exposed to fibrin (0.8 mg/ml, 18 h). A selected panel of genes enhanced by fibrin is shown in figure 1 (the whole set of molecules is enlisted in online supplementary table 1). Briefly, fibrin induced pro-inflammatory genes, including chemokine ligands CXCL-1, IL-8 and CCL-2, ICAM-1 and vascular adhesion molecule-1. Also increased were regulatory molecules participating in acute phase response and in coagulation, such as bradykinin receptor B1, thrombomodulin and the plasminogen activator inhibitor-1, as well as the negative regulators of NFκB, IκBα and TNFα-induced peptide-3. Interestingly, genes associated with invasiveness, as endothelin 1, platelet derived growth factor and histone deacetylase-9, were found increased by fibrin.

Principal transcripts upregulated by fibrin (0.8 mg/ml, 18 h) in one rheumatoid arthritis synovial fibroblasts (RASF) culture as studied with Affymetrix array techniques. IL, interleukin; CXCL, CCL, chemokine ligand; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; TNFAIP, tumour necrosis factor α-induced peptide; COX, cyclooxygenase; NFκB, nuclear factor κ B; SOD, superoxide dismutase; IκB, inhibitor of NFκB; PAI, plasminogen activator inhibitor; DENN/MADD, differentially expressed in neoplastic and normal cells/MAPK activating death domain; HDAC, histone deacetylase; PDGF, platelet derived growth factor; FGF, fibroblast growth factor. Fold-increase of each molecule versus mRNA of untreated cells is shown in brackets.
Fibrin induces pro-inflammatory cytokines in RASF
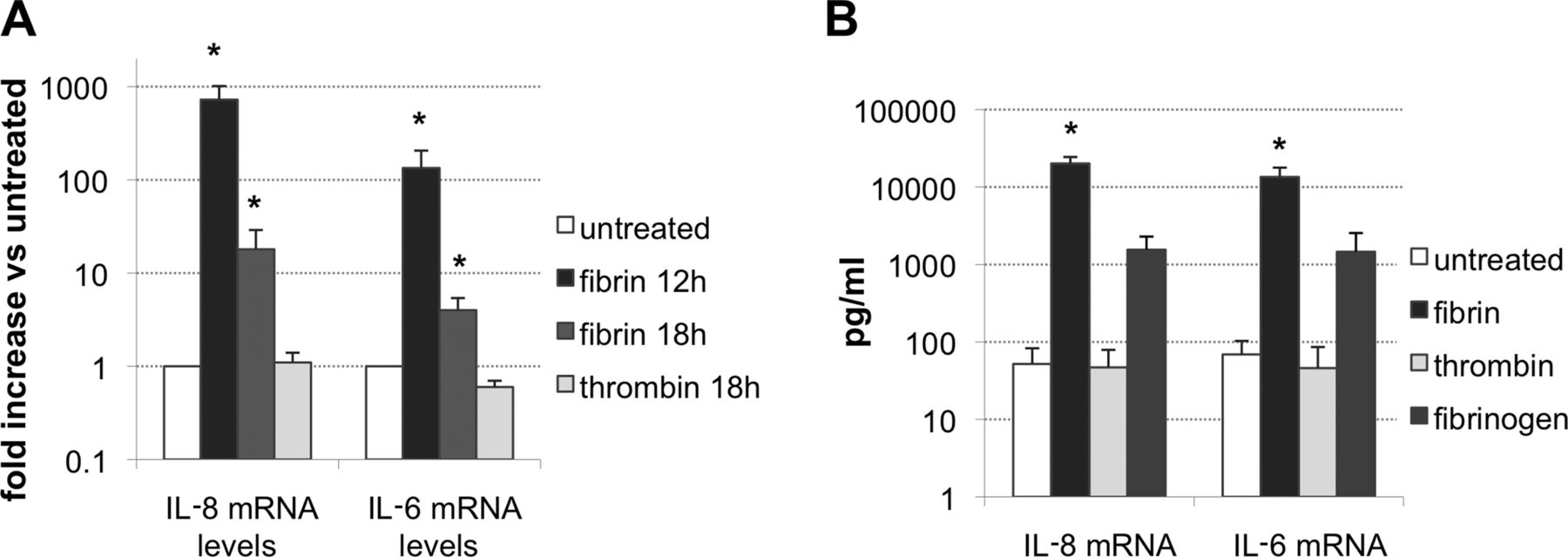
From the target genes identified with the microarrays, we focused our studies on IL-8 and IL-6, and could confirm a substantial induction of both cytokines by fibrin. Fibrin (0.8 mg/ml) elicited a 725 (±290)-fold upregulation of the IL-8 transcript at 12 h (n=6, p<0.05) and an 18 (±11)-fold increase at 18 h (p<0.05, n=6) compared with baseline levels. IL-6 gene expression was 134 (±72)-fold increased at 12 h (p<0.05, n=6) and four (±1.4) fold at 18 h (p<0.05, n=6) (figure 2A).
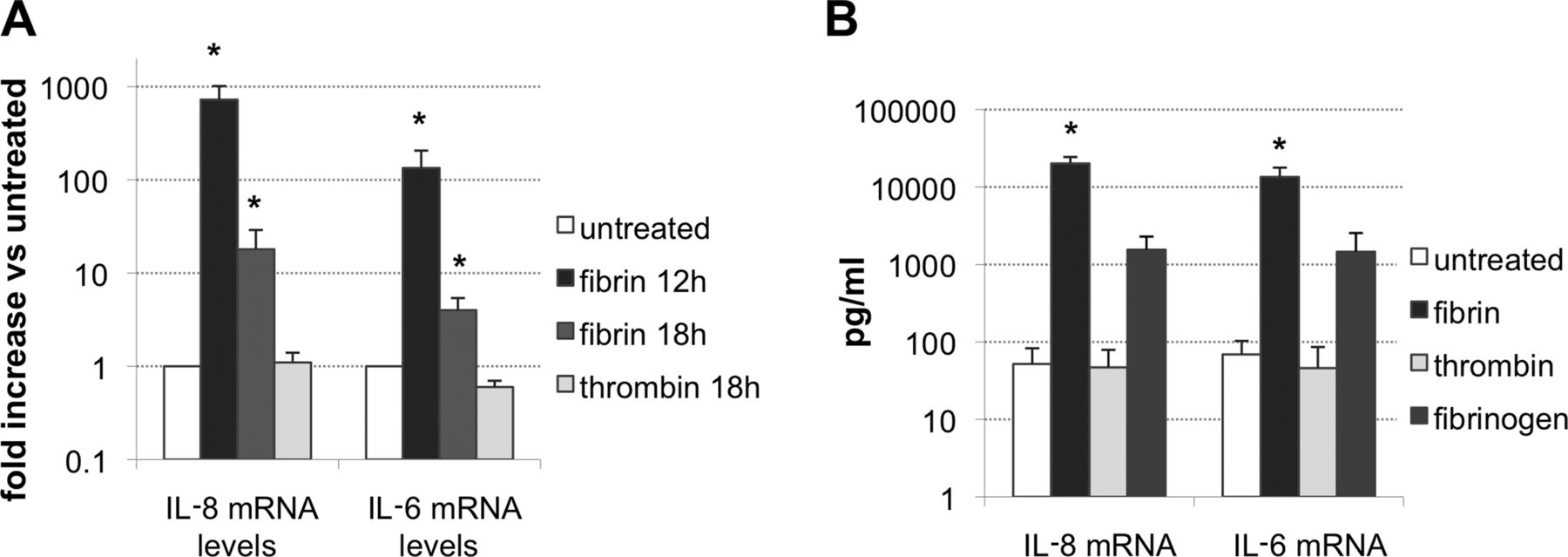
(A) Gene expression of IL-8 and IL-6 in rheumatoid arthritis synovial fibroblasts incubated with 0.8 mg/ml fibrin for 12 or 18 h (n=6) or with 0.75 IU/ml thrombin for 18 h (n=3). Results are shown as relative to mRNA levels of untreated cells, *p<0.05. (B) Levels of interleukin (IL)-8 and IL-6 determined by ELISA in the supernatant fractions of untreated cells, or at 15 h of stimulation with 0.8 mg/ml fibrin (n=6), 0.8 mg/ml fibrinogen or 0.75 IU/ml thrombin (n=4). *p<0.05 Versus untreated.
Thrombin is required for fibrin clotting and might trigger cell responses through the binding of protease-associated receptors.28 As shown in the figure, thrombin (0.75 IU/ml, 18 h) did not elicit changes in mRNA levels of the cytokines compared with non-stimulated cells. IL-8 mRNA relative expression compared with untreated cells was 1.1 (±0.3), while that of IL-6 mRNA was 0.6 (±0.1).
The induction of pro-inflammatory cytokines by fibrin was confirmed on the protein level. Exposure of RASF to 0.8 mg/ml fibrin for 15 h led to a significant increase in the levels of IL-8 (52±31 pg/ml to 20.1±4.3×103 pg/ml, p<0.05, n=6) and IL-6 (69±34 pg/ml to 13.5±4.3×103 pg/ml, p<0.05, n=6) in culture supernatants compared with non-stimulated controls (figure 2B). Incubation with thrombin did not elicit any change in baseline levels, while fibrinogen increased levels of IL-8 to 1.5 (±0.7)×103 pg/ml, and IL-6 to 1.5 (±1.1)×103 pg/ml (n=4), although to a lesser extent than fibrin (p=0.07) (figure 2B).
In additional experiments, we could show upregulation of both gene expression and peptide release to supernatants, at 0.3, 0.5 and 0.8 mg/ml fibrin (n=3, see online supplementary figure S3).
Controls performed excluded that the effects of fibrin were due to LPS contamination of the reagents (see online supplementary figure S4).
TLR4 participates in the pro-inflammatory response of RASF to fibrin
We next tested whether this effect of fibrin could depend on the activation of TLR4. Preincubation of cells with TLR4 blocking antibodies reduced the stimulatory effect of fibrin on IL-8 gene expression from 8.4 (±1.7) to 3.5 (±0.3)-fold (p<0.05, n=5). Likewise, levels of IL-6 mRNA dropped from 5.3 (±1.8) to 2.2 (±0.3)-fold (p<0.05, n=5) (figure 3A). Incubation of cells with an isotype control IgG resulted in negligible differences as compared with the effect drawn by fibrin (n=2, not shown).

(A) Effect of blocking toll-like receptor (TLR)4 on the gene expression of interleukin (IL)-8 and IL-6 induced by fibrin. Murine antihuman TLR4 blocking antibodies (10 μg/ml) were added 90 min before fibrin stimulation. The graph shows mRNA levels of the cytokines at 18 h of incubation, as relative to untreated samples (n=5, *p<0.05). (B) Protein levels of IL-8 and IL-6 in supernatants at 15 h of exposure to 0.8 mg/ml fibrin, with or without prior incubation with antihuman TLR4 blocking antibodies (10 μg/ml) (n=5). (C) Gene expression of IL-8 and IL-6 in cells with selective suppression of TLR2, TLR4 or intercellular adhesion molecule (ICAM)-1 and stimulated with 0.8 mg/ml fibrin for 12 h. Results are shown as relative to mRNA levels of mock transfects (*p<0.05). (D) Levels of IL-8 and IL-6 in supernatants of cells with selective suppression of TLR2, TLR4 or ICAM-1, and stimulated with 0.8 mg/ml fibrin for 18 h. Results are compared with fibrin-stimulated mock-transfected cells (*p<0.05).
Addition of TLR4 blocking antibodies also resulted in a reduction, albeit not significant, of IL-8 levels in supernatants from 27 (±7.9)×103 to 17.7 (±5.6) × 103 pg/ml (p=0.08, n=5) and of IL-6 from 10.7 (±6.7)×103 to 8.1 (±4.9)×103 ng/ml (p=0.08, n=5) (figure 3B).
These results suggested the participation of TLR4 in fibrin-mediated actions in RASF, as it was previously shown in macrophages.11 ,12 In order to confirm this pathway, we studied the effect of fibrin on IL-8 and IL-6 expression in TLR4 k.d. cells, as compared with mock transfects. In these experiments, we also explored the participation of two additional putative receptors of fibrin(ogen), TLR2 and ICAM-1.
Suppression of TLR4 resulted in a significant decrease of 53 (±8)% in IL-8 (p<0.05, n=6) and 61 (±13)% in IL-6 (p<0.05, n=5) mRNA levels, as compared with control transfects (figure 3C). In contrast, the gene expression of both cytokines was unchanged in TLR2 k.d. cells, which showed IL-8 mRNA levels of 89 (±16)% (n=6, NS), and IL-6 mRNA levels of 84 (±18)% (n=5, NS) as compared with mock transfects, as also in ICAM-1 k.d. cells, which evoked a relative expression of 103 (±2)% and 80 (±23)% respectively (n=4) as compared with mock transfects (figure 3C).
There was also a reduction in levels of both cytokines at supernatants in TLR4 negative transfects (figure 3D). IL-8 levels dropped from 13 (±3)×104 pg/ml found in mock-transfected cells to 4 (±1) × 104 pg/ml in TLR4 k.d. cells (p<0.05, n=5). In contrast, IL-8 levels in supernatants from TLR2 k.d. cells were 12 (±3)×104 pg/ml (NS, n=5) and those of ICAM-1 k.d. cells were 14 (±3)×104 pg/ml (NS, n=4). Levels of IL-6 were lowered by TLR4 suppression from 13 (±5)×103 pg/ml to 3 (±2)×103 pg/ml (p<0.05, n=6) as compared with control transfects. Transduction of cells with either TLR2 or ICAM-1 siRNA did not modify supernatant levels of IL-6, which were 12 (±5) × 103 pg/ml (n=6) and 11 (±5)×103 pg/ml (n=4), respectively (figure 3D).
Fibrin activates NFκB in RASF
Engagement of TLR4 can lead to the activation of NFκB, which subsequently triggers the expression of pro-inflammatory genes. With confocal microscopy, we could observe translocation to the nucleus of the p65 component of NFκB at 30 and 120 min of incubation with 0.8 mg/ml fibrin (n=3) (figure 4A). In contrast, the NFκB inhibitor, IκBα, remained in a cytoplasmic location and its signal tended to fade over time (figure 4A). These changes are consistent with NFκB activation upon fibrin stimulation in RASF. In parallel, we could show shuttling of phosphorylated IκBα and of IκBβ from the nucleus to the cytoplasm, that is, in an opposite direction to p65, at 30 and 120 min of stimulation with fibrin (n=2, see online supplementary figures S5 and S6).

(A) Immunodetection of p65 and IκBα in resting RA synovial fibroblasts (0’), and at 30 and 120 min stimulation with fibrin (0.8 mg/ml), as analysed with confocal microscopy, at 400× magnification plus 2× zoom. Arrows point to p65 positive and inhibitor of nuclear factor κ B (IκB) negative cell nuclei indicating NFκB activation upon incubation with fibrin. At 120 min, most of the p65 immunoreactivity was found inside the nuclei, as highlighted in the merged images. (B) Electrophoretic mobility-shift assay performed in nuclear extracts from cells at increasing times of exposure to fibrin. The extracts were incubated with a labelled consensus NFκB probe. An increase in the specific binding was found at 90 min of incubation with fibrin, and persisted at 4 h of incubation. Addition of the unlabelled probe to the 90 min mixture reaction resulted in the displacement of the binding (C mark), confirming specificity of the band pattern. Preincubation of cells with 10 μg/ml toll-like receptor 4 blocking antibodies was shown to reduce the specific NFκB complexes in nuclear extracts of cells exposed to fibrin (T mark).
We further confirmed the presence of NFκB specific complexes in nuclear extracts from cells exposed to fibrin using electrophoretic mobility-shift assay (n=1). An increased binding to the NFκB specific probe was clearly evident at 90 min and at 4 h of stimulation with fibrin (figure 4B). The specific binding was competed by adding unlabelled probe to the reaction (figure 4B). Moreover, the NFκB specific signal was reduced in cells preincubated with blocking anti-TLR4 antibodies (figure 4B).
Citrullination of fibrin amplifies the pro-inflammatory effect of fibrin in RASF
Polymerised fibrin (0.8 mg/ml) was incubated with PAD2 or PAD4 for 2, 4 or 15 h (see online supplementary file S1). After treatment with the deiminating enzymes, there were specific citrulline-enriched peptide sites in fibrin α and β chains consistent with previous reports (figure 5A).29 Optimal citrullination efficiency was found at 4 h incubation with 18 µg/ml PAD2 (enzyme to substrate molar ratio of 1 : 10) and with 41 µg/ml PAD4 (enzyme to substrate molar ratio of 1 : 5), and these were established as working conditions for our experiments. Nonetheless, PAD4 was less efficient than PAD2 in the modification rate of both fibrinogen and fibrin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The citrullination reaction was carried out in equal amounts of fibrinogen and fibrin with recombinant human peptydilarginine deiminase (PAD) 2 and 4. The upper panels show Coomassie staining of lysates obtained from native fibrinogen or fibrin (0) or from the molecules after incubation for 4 h with PAD2 or PAD4 at 1 : 10 and 1 : 5 enzyme to substrate molar ratios, respectively, and subjected to SDS-PAGE. At the lower panels, transferred proteins were incubated with antibodies against citrulline-modified peptides. (B) Gene expression levels of interleukin (IL)-8 and IL-6 in RA synovial fibroblasts incubated with PAD2- or PAD4-citrullinated fibrin for 18 h as compared with levels in cells incubated with native fibrin (n=5, *p<0.05). (C) Levels of IL-8 and IL-6 extracted from non-citrullinated, PAD2 and PAD4-modified fibrin clots after incubation with cells for 24 h (n=5, *p<0.05 vs native fibrin).
We confirmed that these citrullination conditions did not reduce cell viability up to a period of 24 h of incubation (see online supplementary figure S7).
We next compared the effect of PAD2 and PAD4-modified fibrin to the effect of non-deiminated fibrin on the upregulation of IL-8 and IL-6. At 18 h of incubation, PAD2-citrullinated fibrin resulted in a 200 (±190)-fold increase in IL-8 mRNA (p<0.05, n=5) and an 8.5 (±5)-fold increase in IL-6 mRNA (p<0.05, n=5) compared with non-citrullinated fibrin (figure 5B). Levels of IL-8 mRNA yielded by PAD4-citrullinated fibrin were 115 (±80)-fold higher than those of non-citrullinated fibrin (NS, n=5), while IL-6 mRNA levels were 12 (±6)-fold increased (NS, n=5) (figure 5B).
At 24 h of incubation, cytokines released to clot homogenates from the cell cultures were higher in citrullinated than in non-modified fibrin (figure 5C). Non-citrullinated fibrin contained 467 (±240) pg/ml IL-8 and 445 (±146) pg/ml IL-6, PAD2-modified fibrin had 912 (±280) pg/ml IL-8 (p<0.05, n=5) and 772 (±174) pg/ml IL-6 (p<0.05, n=5) and PAD4-modified fibrin showed 841 (±370) pg/ml IL-8 (p<0.05, n=5) and 613 (±155) pg/ml IL-6 (NS, n=5).
In control experiments, we excluded that LPS contamination of PAD2 could account for the effect found (see online supplementary figure S4).
Discussion
In this study, we demonstrate the pro-inflammatory effect of fibrin in RASF through the induction of IL-6 and IL-8, two cytokines of pathogenic interest in RA. Fibrinogen has been shown to promote leucocyte transmigration thereby exerting pro-inflammatory actions. Additional cell types, including synovial fibroblasts, have been found to release chemokines upon stimulation with fibrinogen.30 However, activation of extravascular haemostasis in RA joints puts synovial cells directly into contact with fibrin, the clotted macromolecule. This fact prompted us to explore the relevance of the interaction between fibrin clots and RASF.
Our data show that fibrin displays a stronger activity than fibrinogen in the induction the cytokines. Due to its insolubility, fibrin is difficult to remove from joints and its persistency increases the likelihood of undergoing post-translational modifications and becoming immunogenic.31 Compared with fibrinogen, insoluble clots could result in steadier contacts with target cells. In this regard, monocytes/macrophages show higher binding affinity to fibrin, and also qualitative differences in receptor binding have been described.32 Interestingly, endothelial cells express IL-8 in response to fibrin and not to fibrinogen.27 In colon carcinoma cells, fibrin, but not fibrinogen, was shown to bind P selectin and CD44, and to mediate platelet aggregation.33 Similarly, our findings indicate that immobilisation of fibrinogen into fibrin inside joints is a key event amplifying mechanisms of disease perpetuation.
Along with the cytokines studied, the whole-genome expression assay showed the upregulation by fibrin of a variety of NFκB-responsive molecules (figure 1, see online supplementary file S2).34 ,35 Although these results would need further confirmation, they led us to focus our studies on the TLR4 pathway, which we confirmed to mediate RASF pro-inflammatory response to fibrin. TLR are fully expressed in macrophages, but their presence in RASF has been described.36 Our work suggests that fibrin, as also found with tenascin C,37 acts as DAMP in these cells.
TLR characteristically act in cooperation with other cell surface molecules. The availability of coreceptors could determine the final output, and lead to cell-dependent, and perhaps disease-associated, variations in responses to DAMPs. In this regard, distinct patterns of activation have been described in cells exposed to fibrinogen,38 and also TLR4-drawn responses seem to vary with the engagement of different DAMPs. Of note, TNFα, an end-product of fibrinogen-dependent activation in macrophages,12 ,39 was not found induced by fibrin in our experiments (data not shown). Also in contrast to other studies,40 ,41 we cannot conclude that either TLR2 or ICAM-1 participate in the activation of RASF drawn by fibrin. It remains to be studied whether additional receptors, such as integrins or selectins, can modulate the effect herein described. Differences in cell responses to TLR can derive from signalling pathways downstream receptor engagement. Our results support that canonical activation of NFκB, probably following a MyD88/Mal-dependent pathway, was involved in RASF response to fibrin, as found in other cell types.15 ,42
An interesting subject to study was how citrullination affects fibrin activation of RASF. Some immune mechanisms have been claimed to bridge citrullination of autoantigens and RA severity, including a costimulatory role of citrullinated fibrinogen-containing immune complexes.12 On the other hand, a correlation between the amount of citrullinated fibrin and the development of ACF or the erosive tendency has not been found. However, an amplification of cell activation such as the one here described suggests there could exist a relationship between citrullination of fibrin and severity. In the same way, citrullinated vimentin was shown recently to activate proliferation and expression of RANKL in synovial fibroblasts, whereas the native protein did not.43 The increase in fibrin-evoked responses found with its modification by PAD could derive from changes in the affinity to the TLR4 binding region44 ,45 or from a higher resistance of the modified protein to plasmin cleavage, since both mechanisms have been described.19 ,46
Both PAD2 and PAD4 deiminating activities have been associated with fibrin modification in RA joints.47 In our conditions, PAD4 was less efficient in the transformation of fibrin than PAD2, and resulted in a less consistent upregulation of IL-6 and IL-8. Our findings are in agreement with previous work showing a relative resistance of fibrin(ogen) to PAD4 in vitro.48 In the same line, a recent work showed that actin citrullination is primarily mediated by PAD2.49 These findings support substrate-dependent specificity in the action of PADs and suggest that PAD2 could be the most active isoform in the citrullinating activity associated with RA. However, it cannot be concluded from in vitro studies how citrullination might take place in vivo.
In summary, our data describe a fibrin-derived targetable pathway upstream NFκB activation of resident cells in RA. Our work adds to the increasing evidence on the role of innate pathways involving endogenous activators in the progression of rheumatoid synovitis. The induction of IL-6 by either native or citrullinated fibrin bridges fibrin deposition and severity of RA. We suggest that both the TLR4-fibrin binding domain and PAD2 activity represent attractive targets for novel therapeutic strategies in RA.
Acknowledgments
Dr Sanchez-Pernaute's work was supported by the Spanish Foundation for Rheumatology (FER) and the Conchita Rabago Foundation (FCR). Hanna Maciejewska-Rodrigues is supported by an EU Marie-Curie Grant/ZIHP. Antonio Gabucio has a grant from FCR. Astrid Jüngel is supported by ZIHP and the EU project Masterswitch FP7 and Steffen Gay by the IAR Epalanges. The authors thank Maria del Mar G García-Parreño for her excellent technical support in confocal microscopy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table
Footnotes
-
Handling editor Tore K Kvien
-
Contributors All authors contributed to this work, have been given the opportunity to read the submitted manuscript and have given their consent to sign as coauthors.
-
Funding None.
-
Competing interests None.
-
Patient consent Patients signed a consent form to donate the samples for their use in this study.
-
Ethics approval Ethics committee approval was obtained for this study.
-
Provenance and peer review Not commissioned; externally peer reviewed.