Article Text

Abstract
The therapeutic benefits of immune checkpoint inhibitors (ICIs), which enable antitumor immune responses, can be tempered by unwanted immune-related adverse events (irAEs). Treatment recommendations stratified by irAE phenotype and immunohistopathological findings have only recently been proposed and are often based on those used in primary autoimmune diseases, including targeting of specific proinflammatory cytokines with monoclonal antibodies. Increasing evidence supports the use of such antibody-based strategies as effective steroid-sparing treatments, although the therapies themselves may be associated with additional drug toxicities and reduced ICI efficacy. Kinases are key contributors to the adaptive and innate responses that drive primary autoimmune diseases and irAEs. The janus kinase/signal transducer and activator of transcription, Bruton’s tyrosine kinase, and mitogen-activated protein kinase-interacting serine/threonine protein kinases 1 and 2 pathways are also critical to tumor progression and have important roles in cells of the tumor microenvironment. Herein, we review the histopathological, biological, and clinical evidence to support specific monoclonal antibodies and kinase inhibition as management strategies for irAEs.
- immunotherapy
- inflammation
- autoimmunity
- review
- therapies
- investigational
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapy has revolutionized the management of patients with advanced malignancies. Immune checkpoint inhibitors (ICIs) commonly target proteins that negatively regulate the T cell-mediated host immune response to cancer, namely, cytotoxic T lymphocyte protein 4 (CTLA-4), programmed cell death 1 (PD-1), and programmed cell death ligand 1 (PD-L1), thereby enabling immune activation and antitumor response. ICIs have been approved by the Food and Drug Administration (FDA) for a broad range of solid and hematological malignancies.1 In 2019, approximately 36.1% of patients with cancer in the USA were eligible for ICI therapy, and the use of ICIs continues to increase.2 However, ICIs are also associated with a broad spectrum of autoimmune and autoinflammatory adverse events related to immune activation, referred to as immune-related adverse events (irAEs). They may be severe or permanent, significantly impair quality of life, impact treatment efficacy through dose-limiting effects, or even lead to death.3 irAEs can potentially occur in any organ system and have been found by systematic review to impact 89% of patients treated with CTLA-4 inhibitors, 74% of those receiving PD-1/PD-L1 inhibitors, and 90% of patients treated with combination therapy.4
To date, management for moderate to severe irAEs has been mostly empirical, with systemic corticosteroids as first-line therapy and immune modulators adapted from immune-based approaches employed in primary autoimmune diseases as second-line treatments.5 6 Despite their efficacy in acute irAEs, the long-term corticosteroid use required to control some irAEs has significant systemic toxicity. Recommendations stratified by irAE phenotype and immunohistopathological findings have only recently been proposed.7 8 The janus kinase (JAK)/signal transducer and activator of transcription (STAT), Bruton’s tyrosine kinase (BTK), and mitogen-activated protein kinase (MAPK)-interacting serine/threonine protein kinases 1 and 2 (MNK1/2) pathways have been shown to contribute to the adaptive and innate immune responses that underly primary autoimmune disorders and irAEs.9–12 Therefore, targeting of these kinase pathways represents a potential therapeutic strategy for the management of ICI-induced toxicities. Herein, we review the activity of targeting specific cytokines with monoclonal antibodies, as well as assess the evidence for the use of kinase inhibitors, specifically JAK, BTK, and MNK1/2 inhibitors, in irAEs.
Histopathological and biological basis of irAEs
Observation of specific histopathological findings in various affected organ systems suggests that irAEs are triggered by distinct immunopathogenic mechanisms.7 Infiltrates of predominantly lymphocytes may be seen on histopathology of irAEs involving the skin (eg, maculopapular eruption),13 central nervous system,14 kidney,15 gastrointestinal tract,16 17 and musculoskeletal system.18 Importantly, detailed clonal analysis of ICI-mediated colitis revealed expansion of resident CD8+ T cells as well as infiltration of new T cells into the colon.17 These lymphocyte-predominant histopathological changes are suggestive of an upregulation in tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-17, and/or integrins, and thus their stimulated signaling pathways may be involved in the pathogenesis and management of these irAEs. A second histopathological pattern involves mixed innate and lymphoid infiltrates. These may be seen in cutaneous (eg, lichenoid and psoriasiform),13 19 hepatic,20 pulmonary,21 cardiac,22 renal,15 and gastrointestinal16 17 irAEs. Such histological changes indicate that TNF-α, IL-1, IL-6, IL-12/IL-23, and JAK–STAT signaling may be involved in their development. Autoantibody-mediated toxicities include renal,23 rheumatological,24 cutaneous (eg, bullous pemphigoid),25 and central nervous system irAEs,26 and theoretically implicate JAK–STAT and BTK signaling in their pathogenesis. Recognition of these specific histological findings further supports investigation of tailored approaches to management of irAEs. Since these soluble factors stimulate common downstream signaling kinases, targeting of these kinases may prove more effective than blocking any of the individual factors.
Indeed, increased levels of these proinflammatory cytokines have been found at baseline and early during treatment in the sera of patients who developed irAEs, indicating that they may be predictive biomarkers for and/or early markers of irAE onset (see table 1).27–30 Lower baseline levels and greater post-treatment increases in multiple chemokines have also been associated with the onset of irAEs.28 Moreover, production of these soluble factors is generally regulated by kinases, and they in turn can exert their effects through activation of downstream signal transduction pathways (see figure 1). These can include, but are not limited to, the MAPK, JAK–STAT, phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), and BTK pathways. Therefore, one might posit the importance of specific kinases in the development of irAEs and the potential for efficacy of small molecule kinase inhibitors in their treatment.

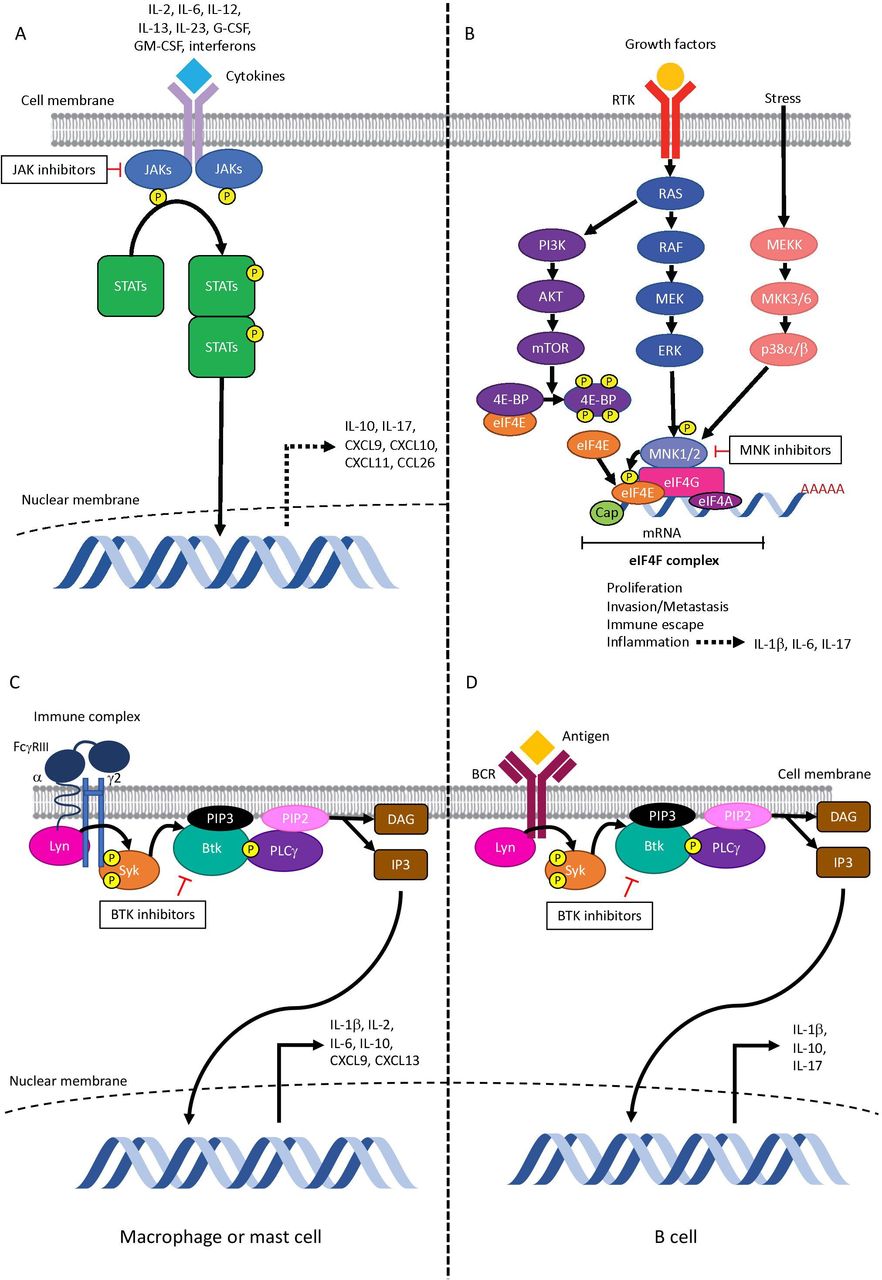
Simplified (A) JAK–STAT pathway, (B) MNK1/2-eIF4E pathway, and BTK signaling in (C) myeloid cells and in (D) B cells. (A) Inflammatory cytokines implicated in irAEs such IL-6, IL-12, IL-23, and interferon signal via JAK–STAT. JAKs phosphorylate tyrosine residues on their cytokine receptors, leading to recruitment and phosphorylation of STATs. Phosphorylated STATs dimerize and translocate to the nucleus, where they promote the transcription of genes encoding proteins with functions in proliferation and inflammation. The JAK–STAT pathway is implicated in the production of cytokines and chemokines involved in toxicities of immune checkpoint inhibitors, including IL-10, IL-17 (by regulatory T cells), CXCL9, CXCL10, CXCL11, and CCL26. (B) MNK1/2 are activated downstream of p38 and ERK MAPK pathways. Type I and type II interferons, which have been implicated in checkpoint inhibitor adverse events, can also activate MNK1/2. PI3K/Akt/mTOR signaling leads to hyperphosphorylation of 4E-BP; eIF4E is released and binds to eIF4G. MNK1/2 bind to eIF4G and phosphorylate eIF4E on serine 209. This increases translation of a subset of mRNAs that promote proliferation, invasion/metastasis, immune escape, and inflammation, including production of cytokines involved in irAEs such as IL-1β, IL-6, and IL-17. (C) In myeloid cells, binding of immune complexes activates FcγRIII signaling. (D) In B cells, antigen binding activates BCR signaling. Lyn and Syk tyrosine kinases phosphorylate BTK, which in turn activates PLCγ leading to generation of DAG and IP3. DAG subsequently activates protein kinase C and causes translocation of transcription factors to the nucleus. Of the cytokines implicated in the pathogenesis of irAEs, BTK is involved in the production of IL-1β and IL-10 by both macrophages and B cells; IL-6, CXCL9, and CXCL13 by macrophages; IL-17 by B cells; and IL-2 by mast cells. BTK, Bruton’s tyrosine kinase; DAG, diacylglycerol; eIF4E, eukaryotic translation initiation factor 4E; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; IP3, inositol-1,4,5-trisphosphate; irAE, immune-related adverse event; JAK, janus kinase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol-3-kinase; PLCγ, phospholipase Cγ; STAT, signal transducer and activator of transcription; 4E-BP, eIF4E-binding protein.
Cytokines and chemokines proposed as biomarkers for irAEs
Targeting specific cytokines and immune cells in cancer, primary autoimmune diseases, and irAEs
The cytokines and immune cell subsets suggested as predictive biomarkers for and/or early markers of irAEs also have roles in the pathogenesis of primary autoimmune diseases and cancer progression. In patients receiving ICI therapy, targeting of these elements may therefore lead to clinical activity against irAEs, tumors, or both (see figure 2).

{kind=link}
{kind=link}
Proposed immunopathologically driven strategies for the management of irAEs. Kinase inhibitors offer the potential to modulate multiple soluble factors that drive irAEs and may therefore offer greater efficacy than targeting individual factors with monoclonal antibodies. JAK inhibition would block signaling of IL-6, IL-12/23, and IL-17, which may be involved in the pathogenesis of primarily lymphocytic and mixed innate and lymphoid toxicities. Both JAK and BTK inhibitors affect B cell function, which may be useful in antibody-mediated irAEs. MNK1/2 blockade may also be an option for predominantly lymphocyte-driven or mixed innate and lymphocyte-driven irAEs, with an added benefit of blocking TNF signaling. Biopsy with immunohistochemical analysis of the affected organ as well as measurement of peripheral blood cytokines and autoantibody levels may be considered to provide additional information to guide treatment decisions. Whether these measures in a single patient will lead to choosing a drug that personalizes a beneficial therapy remains to be formally demonstrated. BTK, Bruton’s tyrosine kinase; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; irAE, immune-related adverse event; JAK, janus kinase; TNF-α, tumor necrosis factor-α.
Interleukin-1
IL-1 has an important role in the acute phase of inflammation. IL-1β is abundant in the tumor microenvironment. It stimulates tumor-associated macrophages, myeloid-derived suppressor cells (MDSCs), and expression of PD-L1 in tumor cells, thus enhancing tumor progression.31 In a preclinical model of melanoma, inhibition of IL-1α signaling led to improved ICI activity through elimination of MDSCs,32 and trials of combined IL-1 inhibition and checkpoint blockade are ongoing in lung cancer (NCT03968419 and NCT03631199). IL-1α also mediates innate and acquired resistance to immunotherapy in melanoma.32 Anakinra is a recombinant IL-1 receptor antagonist that is approved for the treatment of rheumatoid arthritis and various autoinflammatory diseases. In addition to treatment of autoinflammatory conditions, rilonacept (anti-IL-1α and IL-1β), and canakinumab (anti-IL-1β) are approved for recurrent pericarditis and for Still’s disease, respectively. Although IL-1β has been implicated in the pathogenesis of irAEs,27 therapeutic targeting in immune-related toxicities has yet to be investigated. IL-1 blockade has been suggested as a therapeutic approach in rheumatological, neurological, gastrointestinal, respiratory, cardiac, and cutaneous toxicities.8
Interleukin-6
IL-6 is a pleiomorphic cytokine involved in inflammation. It promotes tumor growth, invasion, and metastasis by various mechanisms, including oncogenic feed-forward signaling loops, inhibition of dendritic cell activation/maturation, and increased MDSC survival.33 Tocilizumab and siltuximab are monoclonal antibodies that target IL-6 receptor and IL-6, respectively. Despite promising results of preclinical studies, siltuximab was not associated with objective tumor responses in phase I and II trials involving patients with solid organ malignancies.34 Clinical trials of tocilizumab in multiple cancer types are ongoing (NCT04524871, NCT02767557, NCT03135171 and NCT04940299). Tocilizumab is approved for rheumatoid arthritis, giant cell arteritis, and chimeric antigen receptor T cell-induced cytokine-release syndrome, while siltuximab is approved for Castleman disease. Targeting of IL-6 has been suggested as an approach in severe irAEs during the acute phase, as well as severe or refractory arthritis, myocarditis, vasculitis, pneumonitis, myasthenia gravis, dermatitis, etc.8 13 In a retrospective study, tocilizumab led to clinical improvement in irAEs in 79.4% of patients with multiple subtypes, although no significant impact on survival was observed.35 IL-6 blockade was thus suggested as an alternate therapy for steroid-refractory irAEs.35
Interleukin-12 and interleukin-23
IL-12 and IL-23 also have important functions in the regulation of tissue inflammation, including in the tumor microenvironment. In the tumor microenvironment, IL-23 exerts pro-oncogenic activity particularly by upregulating the immunosuppressive tumor-infiltrating regulatory T cells (Tregs), in part via IL-23 receptor-dependent STAT3 activation.36 STAT3 also reduces gene expression of IL-12/p35 in myeloid cells in the tumor microenvironment,36 thereby inhibiting IL-12-mediated anticancer immunity and further promoting tumor progression. Although modulation of the IL-12/23 axis has been associated with a theoretical risk of tumor promotion in preclinical studies,37 clinical trials have not demonstrated an increased malignancy risk in treated patients.38 However, the impact of IL-12 inhibition on productive antitumor responses to immunotherapy is unclear. Theoretically, blocking IL-12 through the p40 subunit may decrease skewing of newly recruited naïve T cells towards a Th1 axis, with a resultant reduction in the interferon-γ (IFN-γ) signaling critical to effective responses to immunotherapy.39 Ustekinumab is a fully human monoclonal antibody targeting the p40 subunit common to IL-12 and IL-23. It is approved for the treatment of plaque psoriasis, psoriatic arthritis, and inflammatory bowel disease (IBD), and is recommended as first-line therapy in patients with psoriasis who have a history of malignancy. Guselkumab, which targets the IL-23α subunit, is also approved for patients with plaque psoriasis and psoriatic arthritis. IL-12 has been implicated in the development of irAEs.27 Thomas et al reported successful use of ustekinumab in two cases of refractory immune-mediated colitis, with stable cancer response.40 In addition, ustekinumab and guselkumab have been used with moderate to significant improvement in psoriasiform irAEs and maintained antitumor efficacy.41 Targeting the IL-12/23 inflammatory pathway has also been suggested as a therapeutic option in severe or refractory immune-related arthritis.8
Interleukin-17
IL-17 is a proinflammatory cytokine that is mainly produced by T helper 17 cells, although it may also be generated by Tregs via JAK signaling.42 IL-17 is critical to host defense against various infectious diseases and to the pathogenesis of diverse autoimmune conditions, including psoriasis, rheumatoid arthritis, and IBD.43 Furthermore, IL-17 has been implicated in the development of ICI-related psoriasiform dermatitis,44 45 pneumonitis,46 and myocarditis.47 To date, there have been three case reports of use of secukinumab, an anti-IL-17A monoclonal antibody, in irAEs. Psoriasiform skin toxicity was successfully treated in all three patients (exacerbation of pre-existing psoriasis in two, de novo psoriasiform dermatitis in one)44 45 48 and ICI-associated colitis in another patient.45 Although continued response to ICI therapy was seen in two of these patients,44 48 the third patient experienced loss of antitumor efficacy.45 Inhibition of IL-17 disrupts the balance between its protumor and antitumor effects in the tumor microenvironment,43 which may affect the activity of ICIs. Despite this, IL-17 inhibition has also been shown to increase ICI sensitivity.49
Granulocyte-macrophage colony-stimulating factor (GM-CSF)
GM-CSF is a hematopoietic growth factor that triggers proliferation and differentiation of neutrophils, macrophages/monocytes, and myeloid-derived dendritic cells from hematopoietic progenitor cells (reviewed in Tarhini et al50). Sargramostim, a synthetic form of GM-CSF, is approved by the FDA for bone marrow stimulation in the context of bone marrow transplantation, radiotherapy, and in conjunction with treatment for several leukemias. GM-CSF enhances dendritic cell activation for antigen presentation and potentiates lymphocyte antitumor functions (reviewed in Tarhini et al50). Interestingly, in a phase II clinical trial of patients with unresectable stage III or IV melanoma, addition of sargramostim to ipilimumab was associated with lower toxicity, particularly gastrointestinal and pulmonary irAEs, and longer overall survival compared with ipilimumab alone.51 This signals the potential for prophylactic GM-CSF to reduce ipilimumab toxicity. However, the role of GM-CSF in the treatment of active gastrointestinal or pulmonary irAEs has not been investigated, and assessment of the potential of GM-CSF to improve efficacy and/or reduce toxicity of other therapeutic regimens is currently under way (NCT04703426).
B cell targeted therapy
B cells have emerged as having possible roles in the pathogenesis of irAEs, namely, through autoantibody production, antigen presentation, T cell activation, and cytokine release.52 Das et al demonstrated that an increase in the CD21-lo B cell subset (primarily memory cells) and plasmablasts, and a decline in circulating B cells after the first ICI cycle were associated with a higher risk of irAEs.52 Thus, changes in circulating B cells may be an early marker for irAEs. Interestingly, pre-existing advanced B cell differentiation status (larger eBm5/Bm5 memory cell subset, low levels of early differentiated Bm2s) was reported as a negative predictor of overall survival in patients with melanoma.53 Successful targeting of B cells using rituximab, a monoclonal antibody directed against CD20 on B cells, has been reported in immune-related myasthenia gravis, encephalitis, renal vasculitis, and bullous pemphigoid.54–56 Importantly, B cell depletion or absence does not impede antitumor efficacy of PD-1 inhibitors in murine cancer models,57 and anti-CD20 therapy led to clinical benefit in a pilot trial and case series of patients with heavily pretreated, advanced melanoma.58 A clinical trial of rituximab plus combined immune checkpoint inhibition in advanced melanoma is under way (NCT03719131).
Additional potential targets
Inhibition of α4 integrin decreases T cell adhesion, attachment, and migration across the blood–brain (α4β1) and blood–gut barriers (α4β7), where they produce inflammation.59 Vedolizumab, an anti-α4β7 integrin antibody approved for the treatment of refractory IBD, has been used successfully in steroid-dependent or steroid-refractory ICI-related colitis.60 α4 integrin blockade using natalizumab, approved for multiple sclerosis and Crohn’s disease, has been reported in ICI-related encephalitis.26
Other possible strategies for irAEs outside the scope of this review include IgE inhibition, CTLA-4 agonism, and IL-2 modulation.
JAK–STAT inhibition
The JAK and STAT families of proteins play complex and essential roles in cellular processes and mediate downstream signaling of numerous cytokines with resulting diverse physiologic effects. As a result, it is not unexpected that certain JAKs and STATs favor tumorigenesis while others are associated with productive anti-tumor responses.
JAK–STAT pathway in tumorigenesis
Inappropriate hyperactivation of the IL-6–JAK–STAT3 pathway is found in many cancers and is often associated with worse prognosis.33 Constitutive activation of JAK–STAT3 signaling may result from mutations in genes encoding JAK enzymes, as seen in myeloproliferative and hematologic malignancies. Hyperactivation of STAT3 in tumor cells may also occur due to elevated levels of IL-6 in the tumor microenvironment or peripheral blood, loss-of-function mutations involving STAT3 inhibitors, or in a JAK-independent manner via SRC and BCR-ABL1 tyrosine kinases.33
In the tumor microenvironment, IL-6 is produced by the tumor cells, tumor-infiltrating immune cells, and stromal cells.61 IL-6 and JAK–STAT3 signaling drives tumor proliferation and survival, invasiveness and/or metastasis, and angiogenesis while creating a highly immunosuppressive tumor microenvironment (through IL-10, transforming growth factor-β, and vascular endothelial growth factor as well as through PD-L1 expression on tumor cells).61 However, an interesting paradox arises as the JAK–STAT pathway is also required for inflammatory signals leading to antitumor immune activity and thus response to ICIs. Loss-of-function mutations in JAK1 and JAK2 have been identified in patients with melanoma who have innate or acquired resistance to ICIs.62 63 On activation by secretion of IFN-γ by cytotoxic T cells and Th1 cells, JAK–STAT signaling in tumor cells and stroma results in expression of MHC-I, MHC-II, and PD-L1, molecules associated with ICI response.64 These opposing functions of JAK–STAT signaling may be explained by differential phosphorylation of STAT proteins. IFN-γ stimulates phosphorylation of STAT1, STAT3, and STAT5.64 In addition to reducing carcinogenic STAT3 and STAT5 signaling, blockade of JAK1, and to a lesser extent JAK2, downregulates inflammatory STAT1 signaling, thereby potentially reducing antitumor immunity. Together, these findings suggest that careful modulation of the JAK–STAT axis is required to promote tumor suppression while preserving anticancer immune activity.
Rationale for JAK inhibition in irAEs
The JAK–STAT pathway is implicated in adaptive and innate immune responses. JAK–STAT inhibitors have been approved for the treatment of primary autoimmune disorders that serve as prototypes for irAEs: ruxolitinib and baricitinib (JAK1 and JAK2), tofacitinib and peficitinib (JAK1 and JAK3), upadacitinib and oclacitinib (JAK1), and fedratinib (JAK2) (see table 2).
FDA-approved JAK inhibitors
JAK–STAT is induced by signaling of many of the cytokines predictive of irAEs and critical to the immunopathogenesis of their prototypical primary autoimmune disorders and cancer progression, such as IL-6, IL-12, IL-23, and IL-17.9 Although these factors may be targeted individually as discussed previously, modulating the common JAK–STAT signaling pathway may lead to greater therapeutic activity against lymphocyte-driven or mixed innate and lymphoid irAEs, tumors themselves, or both. Other inflammatory cytokines that signal via JAK–STAT may also contribute to inflammatory diseases and predict irAEs, including interferons17 65 and G-CSF.27 To our knowledge, direct targeting of these cytokines has not been reported to date in irAEs. JAK inhibition also impairs production of IL-10,66 CXCL9,67 CXCL10, and CXCL11, which may clear irAEs44 45 and modulate their protumor versus antitumor effects on the tumor microenvironment and tumors themselves. JAK inhibitors may also exert activity in B cell-driven irAEs through suppression of B cell activation/differentiation, inhibition of antibody production, and downregulation of plasmablast development.68
Clinical support for JAK–STAT inhibition
There are recent case reports of treatment of irAEs in patients with JAK–STAT inhibitors, but no large studies have been published.69 70 Direct JAK1/3 inhibition using tofacitinib has been reported in five cases of refractory immune-related colitis.69 70 Four of these patients had failed biologics prior to tofacitinib. Tofacitinib, given at the standard IBD dose (10 mg by mouth two times per day) induced clinical response within days, with long-lasting responses in most patients. Most of the patients had achieved their anticancer response prior to tofacitinib initiation. Consequently, additional long-term studies are needed to understand the clinical impact of tofacitinib on antitumor immune surveillance.
Tofacitinib has also induced clinical remission of irAEs in two patients with ICI-related myocarditis71 and one individual with ICI-associated arthritis,72 with a rapid onset of action and durable responses. These clinical findings also support the further consideration of JAK inhibitors such as tofacitinib in the management algorithm of immune-related colitis, myocarditis, and arthritis.
Balancing potential protumor versus antitumor effects of JAK–STAT inhibition
JAK164 and JAK273 are required for the intact IFN-γ signaling critical to productive antitumor responses to ICIs.39 62 Tumor cell resistance to IFN-γ is a known mechanism of resistance to PD-1/PD-L1 blockade74 through loss of direct antiproliferative and proapoptotic effects on cancer cells, reduced MHC expression/antigen presentation, and decreased recruitment of additional T cells to tumors (reviewed in Nowicki et al75). STAT1 also mediates the expression of antitumor Th1 cytokines, such as IFN-γ and IL-12, which are key to innate and adaptive anticancer responses (reviewed in Yu et al76). Therefore, by blocking Th1 immunity via JAK1, JAK2, or STAT1 inhibition, some patients may experience less robust tumor responses.
However, JAK–STAT inhibitors may also have synergistic anticancer effects with ICIs. Excessive JAK1 signaling in a highly inflammatory environment is an important contributor to cancer immune evasion and development of autoimmunity.77 In addition, resistance to ICIs has been correlated with sustained type I interferon signaling (IFN-α, IFN-β, IFN-ε, and IFN-ω),78 which occurs via JAK1 and JAK3.73 The pro-oncogenic STAT3, and to a lesser extent STAT5 and STAT6 (reviewed in Yu et al76), may also be targeted. Preclinical studies have also shown that JAK2/STAT3 inhibitors decreased in vivo growth of solid tumors.79 Indeed, in preclinical models of non-small cell lung cancer, JAK1/2-STAT1/3 inhibition displayed strong synergistic activity with ICIs in order to overcome treatment resistance.80 These findings are likely due to immunomodulation of the tumor microenvironment and mitigation of protumor inflammatory responses.61 Taken together, JAK1/JAK3 and/or STAT3 inhibition may therefore reduce autoimmunity in the context of irAEs while increasing cancer immune surveillance.
Additional benefits and concerns regarding use of JAK inhibitors
Other benefits of JAK inhibitors include oral administration, versus infusion or injection, and rapid onset of action within days to weeks.9 With respect to toxicities, gastrointestinal perforation, myelosuppression, thrombosis, cardiac conduction delays, and infections have been reported with JAK inhibitors.9 The risk of these potential adverse events will need to be evaluated with respect to use of JAK inhibitors in irAEs.
BTK inhibition
BTK is a non-receptor kinase that is highly expressed in hematopoietic cells. It plays a central role in B cell receptor (BCR) signaling and is required for B cell maturation, differentiation, proliferation, antigen presentation, and survival.81 BTK is also involved in Fc receptor signaling in several myeloid cell populations, including monocytes, macrophages, and neutrophils, which are important components of the tumor microenvironment.
BTK in oncogenesis
BTK signaling is critical for survival and proliferation of B cells in hematologic malignancies and interactions with the tumor microenvironment. BTK is overexpressed in chronic lymphocytic leukemia (CLL) and is required for constitutively activated pathways involved in initiation and survival of CLL cells, including AKT, ERK, and NF-κB.81 In CLL, BTK is also implicated in retention of malignant B cells in their microenvironments.82 Similar BTK-related activation of downstream signaling pathways and tissue microenvironment pro-tumor interactions is seen in certain lymphomas.83
Ectopic BTK expression has also been reported in solid organ cancers. A novel oncogenic BTK isoform that promotes tumor cell survival is overexpressed in breast, ovarian, prostate, and colorectal cancer.84 Elevated expression of BTK in solid tumor cells has been correlated with increased cancer aggressiveness and poor survival.85 Furthermore, BTK blockade results in reduced proliferative activity and increased chemosensitivity in multiple preclinical cancer models,84 although preliminary clinical data from combination approaches with PD-1/PD-L1 inhibition show mild benefit.86
Rationale for BTK inhibition in irAEs
BTK inhibitors may be effective in autoimmune diseases characterized by activation of B cells. Ibrutinib, a first-generation, relatively selective BTK inhibitor, has demonstrated efficacy in preclinical models of autoimmunity,10 11 although most likely through its effects on innate immune cells rather than on B cells. The selective BTK inhibitor evobrutinib is also efficacious in preclinical models of rheumatoid arthritis and systemic lupus erythematosus87 and is in clinical development for these diseases as well as for relapsing multiple sclerosis. Other selective BTK inhibitors are being studied in rheumatoid arthritis and/or multiple sclerosis (NCT04586023, NCT04544449, NCT02626026, NCT04411641, NCT04458051, and NCT0441978). Together, these findings underline the promise of BTK inhibition for primary autoimmune diseases that serve as models for irAEs.
BTK therefore represents a potential therapeutic target in irAEs as it is required to produce many of the cytokines predictive of irAEs and central to the pathogenesis of prototypical primary autoimmune diseases while also providing intrinsic antitumor activity. BTK inhibition has been shown to inhibit IL-1β, IL-6, IL-12, and IL-17 production in preclinical models of autoimmune arthritis10 and psoriasis11 with concomitant improvements in disease activity. Reductions in such cytokines may abrogate irAE subtypes driven by lymphoid or mixed myeloid and lymphoid cells. In addition, given their ability to suppress activation and proliferation of B cells in preclinical models of rheumatoid arthritis,88 targeting the BTK pathway may be helpful in B cell/antibody-mediated irAEs, such as myositis, nephritis, bullous cutaneous, and encephalitis irAEs.
Results of trials on combination therapy of PD-1/PD-L1 inhibitors plus BTK inhibitors
In theory, BTK blockade may improve response to ICIs through inhibition of MDSC-related immune suppression within the tumor microenvironment, thereby facilitating the infiltration of cytotoxic T cells. On the other hand, since BTK is also expressed by tumor-infiltrating B cells that interact with tumor-infiltrating T cells to enhance local immune activation to cancer cells, BTK inhibition may hinder this cooperation and thus decrease ICI efficacy. The net effect of BTK inhibition, in combination with immune checkpoint blockade, is being clarified in preclinical and clinical trials. To date, limited anticancer effects have also been demonstrated in trials of combined BTK and PD-1/PD-L1 inhibition in various metastatic or locally advanced solid tumors.86 89 Phase I and II clinical trials have found that the combination is well tolerated with a similar safety profile to their respective classes,86 89 and frequency of irAEs was not increased with combination therapy. Additional research is required to further characterize the efficacy and role of combined BTK and immune checkpoint blockade in solid tumors and frequency of irAEs.
Concerns regarding use of current BTK inhibitors
The first-generation BTK inhibitor ibrutinib is known to have off-target effects, as it also blocks other tyrosine kinases, including epidermal growth factor receptor, IL-2 inducible kinase, and Tec-family kinases. This off-target binding is often associated with adverse events such as rash, diarrhea, bleeding, infections, atrial fibrillation, and hypertension. Next-generation BTK inhibitors (eg, acalabrutinib and tirabrutinib) have increased selectivity in order to improve efficacy and reduce toxicities. Indeed, lower rates of adverse events have been reported with these agents in clinical trials (NCT01578707, NCT02029443, and NCT03053440). In addition, ibrutinib reverses polarization of Th2 cells to a Th1 phenotype by inhibiting IL-2 inducible kinase,90 which may potentiate autoimmunity. Although this trend has not been seen in preclinical and clinical trials of combined ICI-BTK inhibitor therapy to date, more work is needed to assess risks and benefits in patients with irAEs.
Additional kinase inhibitors: focus on kinases controlling mRNA translation
Aberrant activation of the MNK1/2-eukaryotic translation initiation factor 4E (eIF4E) axis plays a key role in tumorigenesis and cancer progression by selectively facilitating translation of a subset of mRNAs that promote cell proliferation, survival, and metastasis91 (see figure 1B). Phosphorylated eIF4E is the point of convergence of the MAPK/ERK, MAPK/p38, and PI3K/AKT/mTOR pathways, which are hyperactivated in various malignancies.92 Importantly, phosphorylation of eIF4E on serine 209 is catalyzed exclusively by MNK1/2 and increases the oncogenic potential of eIF4E.93 Activity of MNKs and their effectors also regulates immune responses in the tumor microenvironment and has been implicated in driving a protumor microenvironment through survival and tumor infiltration of MDSCs and prometastatic neutrophils.93 94
Given the importance of MNK1/2 in tumor progression, selective inhibitors of the MNK1/2–eIF4E axis have been developed. MNK1/2 blockade has been shown to reduce invasion and metastasis of KIT-mutant melanoma and BRAF V600E-mutant melanoma and breast cancer,95 96 and to enhance sensitivity to mTOR inhibitors.97 In recent years, the MNK1/2 inhibitor eFT508 (tomivosertib) has entered clinical trials for treatment of various cancers. The phase II clinical trial of eFT508 with or without PD-L1 inhibition in microsatellite stable colorectal cancer demonstrated strong target engagement and acceptable toxicity, most commonly constipation, diarrhea, fatigue, myalgia, arthralgia, and rash.98 In fact, one patient experienced a partial response, which lasted for 8 months. eFT508 is currently under investigation in several solid tumors. These findings underline the promise of MNK1/2 inhibitors in the management of aggressive malignancies.
Deregulated MNK1/2 signaling has been implicated in autoimmune and autoinflammatory diseases. MNK1/2 are also activated by type I and type II interferons and mediate expression of multiple pro-inflammatory cytokine production, such as IL-6, IL-17, and TNF-α.12 These cytokines are key to the pathogenesis of autoimmune diseases such as colitis, arthritis, and psoriasis and their levels have been correlated with the development of irAEs.27 28 99 Curiously, successful addition of an mTOR inhibitor to PD-1 blockade has been found to promote maintenance of allograft tolerance and continued antitumor efficacy in patients with cancer.100 101 In a patient with melanoma, during anti-PD-1-mTOR inhibitor combination therapy, frequency of activated and cycling CD4+ and CD8+ T cells, eosinophil count, and levels of irAE-related cytokines/chemokines (eg, IL-5, IL-17, and CXCL10) returned to baseline.100 Elevated levels of circulating IFN-γ-producing T cells were maintained on ICI–mTOR inhibitor combination therapy, necessary for maintained antitumor activity of ICIs. Although targeting of the MNK1/2-eIF4E axis in combination with ICI holds potential for direct anticancer effects and modulation of the tumor microenvironment,93 the specific impact of MNK1/2 inhibition on irAEs and on antitumor cells awaits characterization in future work.
Conclusion
ICIs have led to a paradigm shift in cancer care and improved patient outcomes, at the expense of irAEs that can involve any organ. Innovative multidisciplinary irAE boards and machine-learning approaches have the potential to optimize irAE diagnosis and management to identify high-risk patients. Proinflammatory cytokines and their regulating and/or signaling kinases such as JAK, BTK, and MNK1/2 are involved in immune responses in cancer, primary autoimmune diseases, and irAEs. Increasing evidence supports the use of monoclonal antibodies targeting these cytokines as a potential steroid-sparing strategy. However, tumors are heterogeneous and may secrete more than one of the soluble factors discussed in this review, which activate downstream kinases. Kinase inhibitors may offer the opportunity to abrogate downstream signaling of multiple cytokines, accounting for tumor heterogeneity, and may therefore represent an exciting new therapeutic strategy for management of irAEs based on histopathological, biological, and clinical data. Further evaluation through rigorous clinical studies and consensus discussion is required to support their efficacy and future role in treatment algorithms of irAEs.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human subjects.
References
Footnotes
Twitter @delRincon_lab
SVdR and WHM contributed equally.
Contributors Design: M-HHB, SVdR, and WHM; writing: M-HHB, SVdR, and WHM; figures:M-HHB; review and editing: SVdR and WHM. All authors have read and agreed to the submitted version of the manuscript.
Funding This research is funded by the Canadian Institutes of Health Research (grant PJT-162260 to SVdR, and grants MOP-142281 and PJT-156269 to WHM and SVdR) and the Canadian Cancer Society-Emerging Scholar Award (grant number 707140 to SVdR). M-HHB was financed by the Canada Graduate Scholarships Master’s program, Canadian Institutes of Health Research and the Bob Lavoie Research Award, Research Institute of the McGill University Health Centre.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.