Article Text

Abstract
Background and aims A general dysbiosis of the intestinal microbiota has been established in patients with Crohn's disease (CD), but a systematic characterisation of this dysbiosis is lacking. Therefore the composition of the predominant faecal microbiota of patients with CD was studied in comparison with the predominant composition in unaffected controls. Whether dysbiosis is present in relatives of patients CD was also examined.
Methods Focusing on families with at least three members affected with CD, faecal samples of 68 patients with CD, 84 of their unaffected relatives and 55 matched controls were subjected to community fingerprinting of the predominant microbiota using denaturing gradient gel electrophoresis (DGGE). To analyse the DGGE profiles, BioNumerics software and non-parametric statistical analyses (SPSS V.17.0) were used. Observed differences in the predominant microbiota were subsequently confirmed and quantified with real-time PCR.
Results Five bacterial species characterised dysbiosis in CD, namely a decrease in Dialister invisus (p=0.04), an uncharacterised species of Clostridium cluster XIVa (p=0.03), Faecalibacterium prausnitzii (p<1.3×10−5) and Bifidobacterium adolescentis (p=5.4×10−6), and an increase in Ruminococcus gnavus (p=2.1×10−7). Unaffected relatives of patients with CD had less Collinsella aerofaciens (p=0.004) and a member of the Escherichia coli–Shigella group (p=0.01) and more Ruminococcus torques (p=0.02) in their predominant microbiota as compared with healthy subjects.
Conclusion Unaffected relatives of patients with CD have a different composition of their microbiota compared with healthy controls. This dysbiosis is not characterised by lack of butyrate producing-bacteria as observed in CD but suggests a role for microorganisms with mucin degradation capacity.
- Crohn's disease
- dysbiosis
- DGGE
- mucin degradation
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
The role of bacteria in the pathogenesis of Crohn's disease (CD) is well established (clinical presentation and lesions of the disease are localised in areas of highest bacterial exposure, clinical response of patients to diversion of the faecal stream and relapse on restoration or exposure to faecal material, clinical response to antibacterial treatment, absence of disease in germ-free animal models, susceptibility genes for CD are involved in recognition of and defence against microorganisms).
In patients, a general dysbiosis has been described with at the group-specific level lower numbers of members of the Firmicutes phylum and of the Clostridium cluster IV combined with higher numbers of Bacteroides.
Until now, the species-specific finding that has been most convincingly replicated is the lower presence of Faecalibacterium prausnitzii in patients with CD both in faecal samples and in biopsies.
What are the new findings?
We now provide the first detailed description of this dysbiosis in patients with CD.
Based on differences in the predominant faecal microbiota in the largest cohort studied so far, we were able to describe a dysbiosis signature associated with CD, characterised by five bacterial species, namely Dialister invisus, an uncharacterised species of Clostridium cluster XIVa, Faecalibacterium prausnitzii, Bifidobacterium adolescentis and Ruminococcus gnavus.
This dysbiosis signature was markedly characteristic for the disease as it was not observed in unaffected relatives despite a common genetic background and shared nutritional habits.
We demonstrate further for the first time that unaffected relatives of patients with CD also have a different composition of their microbiota compared with healthy controls.
How might it impact on clinical practice in the foreseeable future?
This first detailed description of CD-associated dysbiosis will break new ground in unravelling the role of bacteria in CD.
Furthermore, uncovering this subclinical dysbiosis in unaffected subjects at risk allowed us to hypothesise that enhanced mucin degradation capacity of the intestinal microbiota might be an intermediate step towards CD and disease-associated dysbiosis.
Background and aims
The role of the commensal microbiota in the initiation and perpetuation of the intestinal inflammation in Crohn's disease (CD) is well established.1–5 A general dysbiosis in patients with CD has been described with both culture-dependent and culture-independent techniques,6–8 but whether the observed dysbiosis is either cause or consequence of the disease remains unclear.4 Several studies have reported an overall lower biodiversity in patients with CD. At the group-specific level, lower numbers of members of the Firmicutes phylum and of the Clostridium cluster IV combined with higher numbers of Bacteroides have been reported.9–11 In several studies a proportional abundance of mucosa-associated Escherichia coli in CD has been described, but these findings are self-evidently limited to mucosal samples.12–14 Until now, the species-specific finding that has been most convincingly replicated is the lower presence of Faecalibacterium prausnitzii in patients with CD both in faecal samples and in biopsies.7 15–17 Moreover, the ability to produce butyrate and the anti-inflammatory properties of this species provide a coherent biological explanation for its lower presence in patients with CD.15 A more extended and detailed description of the observed dysbiosis in CD is, however, still lacking.
The abnormal response to commensal intestinal bacteria in CD might be driven by the genetic background of the host, as many of the identified susceptibility genes for CD play a role in bacterial sensing. The first gene identified in CD, NOD2/CARD15, belongs to the family of pattern recognition receptors (PRRs) which are part of the human innate immunity.18 19 PRRs recognise specific bacterial sequences and are responsible for the defence against those organisms. Toll-like receptors (TLRs) were the first class of cellular PRRs identified and are the best known PRRs.20 An association between the functional Asp299Gly polymorphism in TLR-4 and inflammatory bowel disease (IBD) has been demonstrated.21 Also the importance of autophagy in CD became clear as a result of genome-wide association scanning which has identified the autophagy-related 16-like 1 (ATG16L1) gene and immune-related guanosine triphosphatase (IRGM).22 23 To test the impact of the host genotype on the intestinal microbiota, comparisons were made between faecal samples from subjects with differing genetic relatedness, varying from unrelated persons to monozygotic twins.24–26 A higher similarity between samples was found with a higher degree of genetic relatedness. These studies thus indicated a significant impact of the host genotype on the bacterial composition of faecal samples but did not find a clear environmental effect. The interaction of host genotype with the composition of the intestinal microbiota is still poorly understood, but CD twin studies provide evidence that the bacterial composition is more influenced by disease status than by the genetic background of the host.7 17
Although supported by little evidence, the composition of the intestinal microbiota is also likely to be influenced by other factors such as dietary habits and the environment. Both genetic and environmental factors are shared within families, and first-degree relatives of patients with CD are at much higher risk of developing CD as compared with the general population.27 In this regard, we previously reported a 57-fold increase in the incidence of IBD within multiply affected families.28 29
The aim of the current study was to investigate to what extent the predominant faecal microbiota of patients with CD has unique characteristics that discriminates them from healthy subjects with and/or without a shared familial background. Furthermore, we hypothesised that intestinal dysbiosis might be a precursor for CD and, as a consequence, that relatives, or a subgroup of them, also might have asymptomatic intestinal dysbiosis.
Methods
Subjects and study design
Microbial DNA from stool samples of patients with CD was compared with that of their unaffected relatives and healthy subjects from families of comparable generation, gender distribution and family composition for whom the recruitment and matching criteria were previously described.29 Subjects taking antibiotics within 4 weeks before the faecal sampling, following a specific diet, using probiotics or suffering from irritable bowel syndrome (IBS) were excluded from the analyses. A total of 68 patients with CD from 25 families, 84 of their unaffected relatives and 55 healthy subjects from 10 control families were studied (table 1). Four families had only one member affected with CD; 21 families had at least three affected members. Of the 84 unaffected relatives in our cohort, 80 were first-degree (thus primary) relatives of patients with CD (48 siblings, 31 parents of patients with CD and one unaffected subject that had both affected siblings and offspring). The four that did not have first-degree relatives with CD lived in a close relationship with four patients with CD.
Faecal samples used for analysis represented per group
We used large families which resulted overall in balanced numbers of unaffected relatives and patients per family. The median number of unaffected relatives used per family was 3 (IQR 1–4). For patients, the median number per family was also 3 (IQR 2–3). The median ratio of unaffected relatives to patients per family was 1 (IQR 0.67–1.75). All patients were in clinical remission at the time of sampling. The patient characteristics at the time of sampling are summarised in table 2.
Patient characteristics at the time of sampling
The Ethics Committee of the University of Leuven (Belgium) approved the study, and all participants gave informed consent. Faecal samples were collected from each subject and upon collection they were immediately frozen at −80°C and stored for DNA extraction.
Denaturing gradient gel electrophoresis (DGGE) profiling
To identify potential microbial characteristics of CD, an objective comparison by means of a community fingerprinting approach based on DGGE analysis of ribosomal PCR amplicons was performed.30 Therefore, total bacterial DNA was extracted from the faecal samples using the method of Pitcher and colleagues with slight modifications.31 Next, community PCR was conducted using universal primers F357+GC clamp and R518 targeting the hypervariable V3 region of the 16S rRNA gene. The resulting 16S rRNA gene amplicons were analysed with DGGE using a 35–70% gradient as previously described,31 resulting in profiles of the predominant faecal microbiota per subject. On each DGGE gel, a standard reference consisting of an amplicon mix of 12 different bacterial species was included in the middle and at both outer ends for digital gel normalisation and to allow comparison between gels.
Digital processing of DGGE profiles
DGGE profiles were digitally processed with BioNumerics software version 4.6 (Applied Maths, St-Martens-Latem, Belgium) in a multistep procedure following the manufacturer's instructions. After normalisation of the gels, individual bands in each sample lane were marked using the auto search bands option, followed by manual correction if necessary. All profiles were compared using the band matching tool and uncertain bands were included in the position tolerance settings. Allocation of bands to so-called band-classes was performed automatically and again checked manually. Essentially, band-classes were arbitrarily generated in a collective analysis of all profiles by tracing common bands across different sample profiles. This allowed every band in a profile to be assigned to its nearest band-class. A maximum error of 0.5% deviation was applied, which means that a band was only allocated if it was located at a distance of <0.5% of the total length of the profile from the closest band-class. The designation of the band-classes was based on their position on the profile compared with the standard reference. The intensity of a given band-class was expressed with respect to the other band-classes on the same profile.
Comparative analyses of the DGGE profiles
The quantitative information derived from relative band intensities per subject and per band-class was exported as a data matrix. Bacterial groups present in quantities under the detection limit were denoted as zero. Non-parametric tests were applied to compare differences in the complex profiles across groups (table 1). Therefore Mann–Whitney U tests on the intensity per band-class were used to compare the groups in SPSS V.17.0. To correct for multiple testing, all p values were multiplied by the number of band-classes obtained (n=75). Only two-tailed p values <0.05 after correction were considered significant.
DGGE band extraction and sequencing
DGGE bands that differed significantly between groups according to the statistical analysis were purified from the complex faecal samples, before they were sequenced, by excision from the gel. The DNA was eluted from the gel slice into 15 μl of TE buffer by heating the mixture of buffer and gel for 10 min at 65°C. The resulting DNA solution was then amplified again using the universal V3 16S rRNA primers F357+GC clamp and R518. Purification of the bands was facilitated by using DGGE gel expansion where the amplicons were checked by DGGE with an adjusted gradient32 and again excised at least three times until a single band was obtained. The purpose of repeating DGGE until one single band is obtained is only to purify the PCR product before it can be sequenced. In a final DGGE round, purified amplicons were analysed together with the original sample profiles from which they were excised to check visually whether the correct bands were purified.
When purified bands matched with the targeted bands in the sample profiles, amplicons were selected for sequencing using an ABI Prism 3130 Genetic Analyzer (Applied Biosystems, Foster City, California, USA). Sequences were read from both directions with primers F357 (without clamp) and R518, respectively. The sequence was assembled with BioNumerics software. Homology searches of the GenBank DNA database were performed with BLAST search. Based on the BLAST results, reference sequences of phylogenetic neighbour species (up to 90% similarity) were included for clustering analysis using multiple sequence alignment and the average linking method to confirm allocation of the purified band sequences to the most probable species.
Real-time PCR
Bacterial species that differed significantly in the comparative DGGE band-class analyses were quantified with the Applied Biosystems 7500 Fast Real-Time PCR (RT-PCR) System in the fast 7500 mode. Primers (Primer express 3.0 software) that spanned a target region in the V3 region of the 16S rRNA gene species of interest were selected (Supplementary table 1). The RT-PCR was carried out in a 20 μl total reaction mixture using 4 μl of DNA. Calibration curves were constructed using dilutions of genomic DNA from a control strain (Supplementary table 1) for which the number of bacteria was determined by plate counting. The quantitative results of the RT-PCR analyses were based on measurements in triplicate with a maximum variation <0.5 Ct. Amounts of bacterial species were expressed as log10 values per gram wet weight of faeces.
Results
Differences in five bacterial species characterise predominant dysbiosis in CD
In the 207 PCR-DGGE profiles analysed in this study, 75 band-classes were identified. We first compared patients with CD versus all unaffected controls (unaffected relatives and unrelated controls taken together) in search of disease-associated characteristics of the predominant faecal microbiota. After Bonferroni correction for the 75 band-classes, significant differences were observed between the patients with CD and unaffected subjects for seven band-classes. To assign these seven band-classes to a bacterial species or phylotype, DNA was purified per band-class from at least five different samples originating from different patients, unaffected relatives and controls. Purified amplicons were analysed together with the original sample profiles from which they were excised, to check visually whether the correct bands were purified. After purification, band-class 3.72 appeared to be composed of different bands. We thus concluded that this band-class did not represent a single bacterial species and therefore it was not studied further. All other purified bands of the remaining six band-classes matched with the targeted bands in the sample profiles, and these amplicons were sequenced. Within each band-class all generated sequences (length 135 bp, representing 8.7–9.5% of the 16S rRNA gene) were identical. Band-classes were tentatively assigned to a bacterial species or phylotype based on the highest (98–100%) sequence similarity match to GenBank sequences obtained by BLAST analysis (Supplementary table 2). These species assignments were subsequently confirmed by clustering analysis with reference sequences of the closest phylogenetic neighbours. In this way, band-classes 9.68 and 11.51 were both assigned to F prausnitzii, band-class 11.00 was assigned to Ruminococcus gnavus, band-class 14.60 was assigned to Dialister invisus and band-class 16.24 was assigned to Bifidobacterium adolescentis. Based on its phylogenetic position revealed by clustering analysis, band-class 10.79 could be assigned to an uncharacterised species of Clostridium cluster XIVa.
The disproportion of these five bacterial species that characterise the observed differences between the patients with CD and unaffected subjects are represented per band-class in figure 1 and in figure 2 the occurrence in the DGGE profiles is expressed in terms of a percentage.
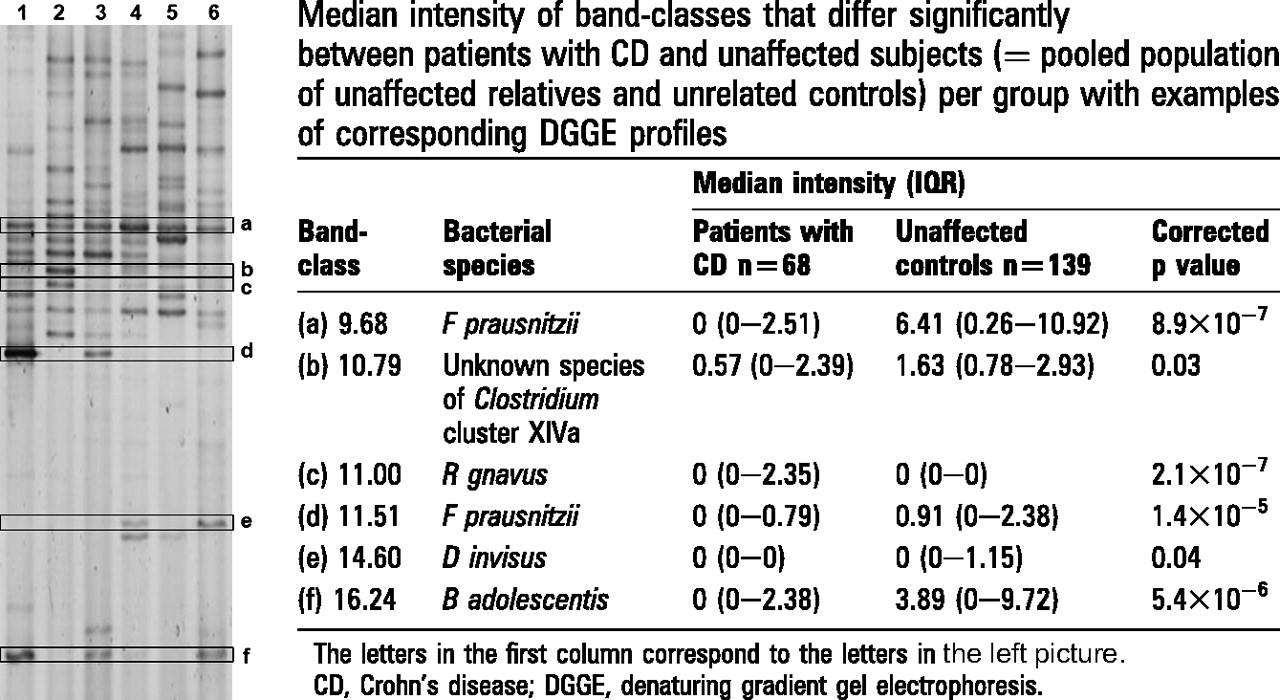
Examples of denaturing gradient gel electrophoresis (DGGE) profiles. The letters next to the picture correspond to the letters in the right table per bandclass. Each of the six lanes shown on the left represents a DGGE profile visualising the predominant faecal microbiota of one subject.
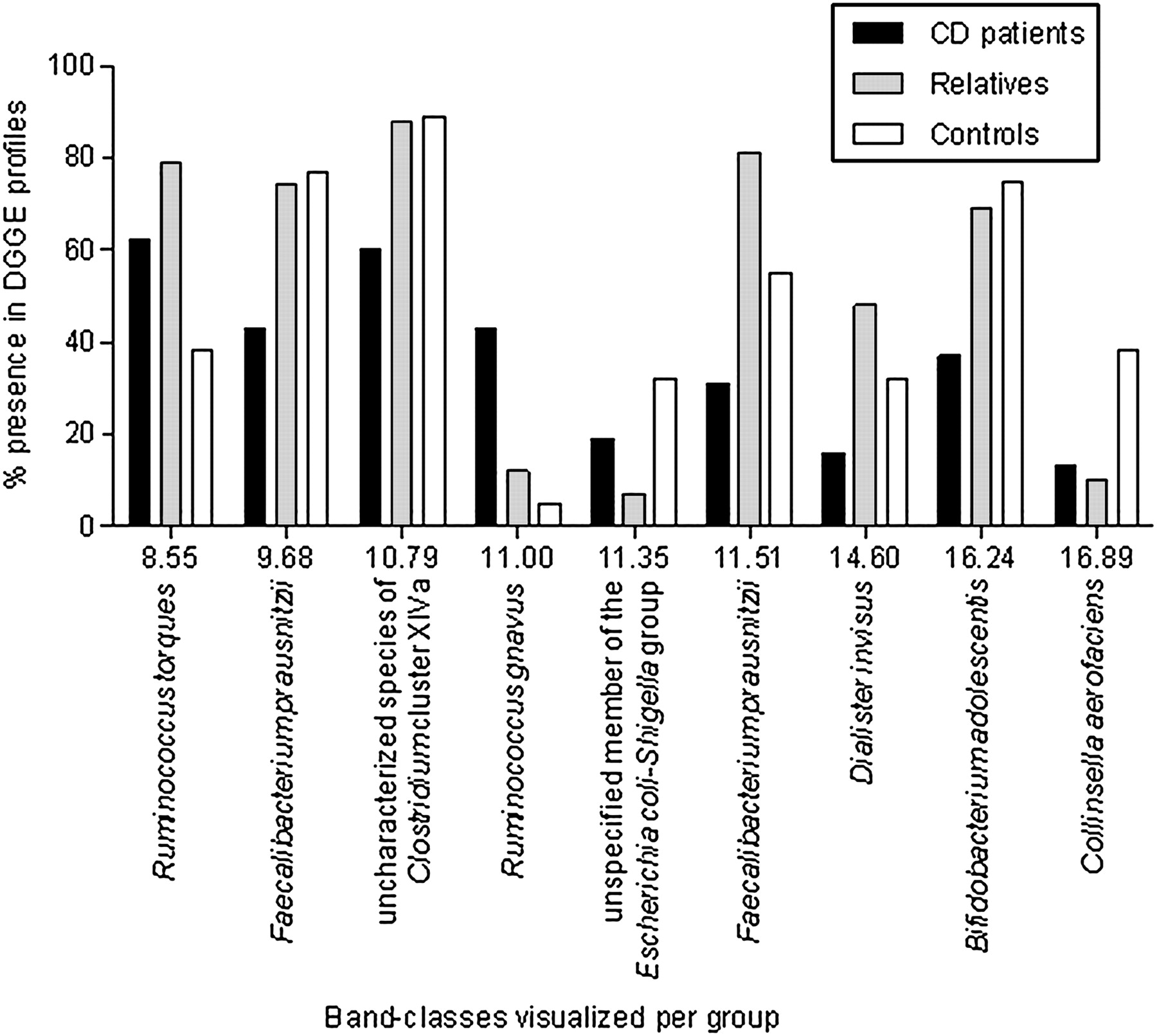
Denaturing gradient gel electrophoresis (DGGE) band-classes that differed in the predominant faecal microbiota of patients with Crohn's disease (CD), their unaffected relatives and control subjects, expressed as a percentage of subjects with the respective band in their predominant profile per band-class and per group. Inclined, the corresponding bacterial species are added.
We also analysed the predominant faecal profiles of the patients with CD according to the clinical phenotypes described in table 2. However, none of the dysbalanced bacterial species was associated with a specific clinical phenotype. Only when comparing predominant microbial profiles of the subgroup of patients with CD that underwent an ileocolonic resection (n=16) prior to sampling with the subgroup of patients that never had an intestinal operation (n=20) was a significant quantitative difference in F prausnitzii revealed (corrected p value=0.002) with even less F prausnitzii in patients with CD after ileocolonic resection.
Influence of the genetic and environmental background on the predominant microbiota
Because the composition of the intestinal microbiota is likely to be influenced by many factors, and both genetic and environmental factors are shared within families, we next compared patients with their unaffected relatives. No differences could be found between patients or unaffected relatives according to the number of affected relatives. In other words, we did not find differences between single incidence families and multiplex families. Therefore, we combined both groups.
However, when comparing the patient group only with their unaffected relatives, all six band-classes except band-class 10.79 (unknown species of Clostridium cluster XIVa) remained significantly different between both groups and no additional discriminative band-classes were identified.
To evaluate the effect of the genetic and environmental background on the observed differences in patients with CD, we then compared the patient group with unrelated control subjects only, to allow a larger potential impact of genetic and environmental factors. All except band-classes 11.51 (F prausnitzii) and 14.60 (D invisus) remained significantly different between both unrelated groups.
Subclinical dysbiosis in asymptomatic relatives of patients with CD
Profiles of unaffected relatives were compared with those of unrelated control subjects in search of asymptomatic intestinal dysbiosis in relatives. Three band-classes differed significantly between both unaffected groups—that is, band-classes 8.55, 11.35 and 16.89 (table 3). Interestingly, the differences observed between unaffected relatives and unrelated control subjects were also observed in patients with CD compared with unrelated controls, but to a lesser extent, and therefore were not significantly different. For the predominant occurrence of the bacterial species that (statistically) differed between unaffected relatives of patients with CD and healthy controls, the values of the unaffected relatives are at one extreme, those of the healthy controls at the other extreme, and those of patients with CD are in between. The prevalence of patients with CD for these band-classes was thus situated in between the prevalence of unrelated controls and that of unaffected relatives (figure 2). After purification and sequencing, BLAST analysis in GenBank revealed 100% similarity of band-class 8.55 to Coprococcus comes, Clostridium nexile and Ruminococcus torques. Band-class 11.35 showed 100% sequence similarity to members of the E coli–Shigella group whereas band-class 16.89 was identified as Collinsella aerofaciens. Subsequent clustering analysis confirmed that band-class 11.35 can be assigned to a member of the E coli–Shigella group, and that band-class 16.89 could be assigned to C aerofaciens. Clustering analysis indicated that R torques rather than C comes or C nexile was phylogenetically the closest species corresponding to band-class 8.55.
Median intensity of band-classes that differed significantly between unaffected relatives of patients with Crohn's disease and unrelated controls
To check for family-associated effects on band-class distribution, we analysed the band-classes using family as the grouping variable and did not find significant differences for the band-classes that we identified as characterising CD-associated dysbiosis or for those characterising subclinical dysbiosis in unaffected relatives.
Quantitative validation of disease-associated differences in predominant microbiota with RT-PCR
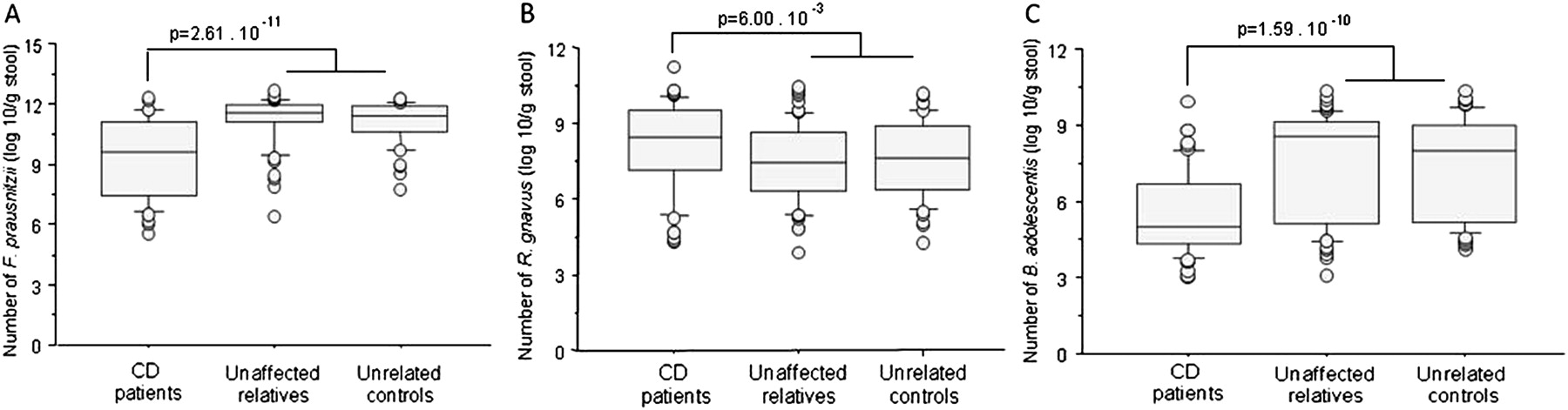
CD-associated predominant dysbiosis was consistently determined by differences in band-classes assigned to F prausnitzii, R gnavus and B adolescentis in the different comparisons made. Therefore, these three species were quantified with RT-PCR. As the quantitative data obtained were not normally distributed (Kolmogorov–Smirnova test statistics: p values <0.023), we performed a non-parametric Mann–Whitney U test to compare the groups. The median number of F prausnitzii was 9.58 log10/g in patients with CD (IQR 7.40–11.07 log10/g) and 11.45 log10/g in unaffected controls (IQR 10.92–11.90 log10/g) (p=2.61×10−11) (figure 3A). The median number of R gnavus was 8.44 log10/g in patients (IQR 7.17–9.51 log10/g) and 7.51 log10/g in unaffected controls (IQR 6.31–8.81 log10/g) (p=6.00×10−3) (figure 3B). The median number of B adolescentis was 5.00 log10/g in patients (IQR 4.31–6.65 log10/g) and 8.34 log10/g in unaffected controls (IQR 5.21–9.14 log10/g) (p=1.59×10−10) (figure 3C). The differences in the quantitative data per group reflect the disproportions identified in the predominant microbiota and provide a quantitative validation of the DGGE band-class analysis results.

{kind=link}
{kind=link}
{kind=link}
Boxplots of the number of (A) F. prausnitzii (B) R. gnavus and (C) B. adolescentis in log10 per g of faeces per group. CD, Crohn's disease.
Discussion
In this study we identified five disproportionate bacterial species that characterise dysbiosis in the predominant faecal microbiota of a large cohort of patients with CD. This dysbiosis signature we found in patients with CD was markedly characteristic for the disease as it was not observed in unaffected relatives despite a common genetic background and shared nutritional habits. The butyrate producer F prausnitzii occurred significantly less in the predominant microbial profiles of patients with CD. In agreement with recent findings of Sokol and co-workers, significantly lower numbers of this species were also detected by RT-PCR in patients with CD as compared with unaffected subjects.33 In contrast, we found that R gnavus appeared more in the predominant faecal microbiota of patients with CD. The higher occurrence of R gnavus in patients with CD has been described previously in mucosal samples of patients with ileal disease.34 We were also able to demonstrate the higher prevalence of this species in faecal samples of patients with CD, both in the predominant profiles and by quantitative PCR. In vitro, R gnavus expresses very high β-glucuronidase activity.35 β-Glucuronidase activity can induce the formation of toxic compounds in the colon which might cause or perpetuate local inflammation. A third bacterial species that clearly differed between patients with CD and unaffected controls was B adolescentis. The lower occurrence of bifidobacteria in general in patients with CD has been described earlier,36 but we have now specifically demonstrated for the first time the lower prevalence of B adolescentis both qualitatively and quantitatively.
To our knowledge, the lower occurrence of D invisus in patients with CD has never been reported before. Interestingly, this species was first isolated from dental root infections,37 but has also been detected as part of the normal intestinal microbiota.38 The sequence of band-class 10.79 was assigned to an uncharacterised species of Clostridium cluster XIVa that was under-represented in the predominant microbial profiles of patients with CD. This sequence showed 100% similarity with the 16S rRNA sequence of strain SS2/1 which was previously found in the colonic microbiota of three healthy adult humans and was classified as a Clostridium indolis-like member of Clostridium cluster XIVa.39 Although this unknown species has not been fully taxonomically characterised, its clear phylogenetic affiliation to butyrate producers of cluster XIV and its lower occurrence in patients with CD can be considered analogous to the observations for F prausnitzii. So far, however, the lower prevalence of this unknown Clostridium species has not been related to CD.
Another major finding in this study is that in unaffected relatives asymptomatic differences in three other bacterial species were also identified. Both an unspecified member of the E coli–Shigella group and C aerofaciens were less prevalent in relatives of patients with CD. Strains of C aerofaciens were previously isolated from patients with colon cancer, ulcerative colitis and CD.40 The lower prevalence of this species in unaffected relatives compared with unrelated controls is a novel finding.
Band-class 8.55, which was found more in relatives of patients with CD, phylogenetically most closely resembled C nexile, R torques and C comes. The latter two commensal species have previously been linked to CD.16 41 A possible role for C comes in the pathogenesis of CD has been suggested based on its interaction with the immune system.42 C comes can activate complement and thereby induce inflammation. Moreover, these bacteria are not ingested by neutrophils and can bind immunoglobulins through their Fc region which prevents phagocytosis of this microorganism.42 For R. torques no role in CD pathophysiology has been suggested so far. Like R gnavus, R torques belongs to Clostridium cluster XIVa, but, in contrast to the other members of this group, R gnavus and R torques are non-butyrate-producing members.43 Moreover, like R gnavus and Candida albicans, R torques is known to degrade gastrointestinal mucin.43 44 For C albicans, the capacity to break down mucus and penetrate the mucin barrier also plays a role in its virulence.44 Degradation of the mucosal barrier might thus enhance bacterial translocation and lead to increased permeability of the gut. C albicans was more frequently isolated from stool samples from patients with CD (44%) and their unaffected relatives (38%) compared with controls (22%) in a cohort comprising similar French families and the same subjects used for the present study.45 As R torques was also more frequently found in relatives of patients with CD, an enhanced mucin degradation capacity of the intestinal microbiota might precede or predispose further to CD.
The intestinal microbiota of patients with CD has been studied with different molecular techniques. Several findings had previously been reported in various publications, but so far these were only partial and/or not very persuasive. In this study DGGE was used as a molecular technique to explore predominant differences in faecal samples of a large cohort of patients with CD, their unaffected relatives and healthy controls. We were able to describe in detail differences among the most abundant members of the faecal microbiota and to confirm and integrate previous findings. In our opinion, large cohorts are needed to study complex communities and, despite being laborious, DGGE is a robust technique and highly valuable as an exploratory tool.
The recruitment of the patients with CD through a tertiary referral centre resulted in a cohort of patients with complicated CD. Moreover, the specific design of the study, using both related controls at increased risk for CD (mainly from multiplex families) and independent control subjects, precluded us from also powering the study for potential effects of disease phenotype or treatment. Subanalyses only revealed that patients with CD might have even less faecal F prausnitzii after ileocolonic resection compared with those that were never operated on. As the group of patients in which we could evaluate this was very small, whether this is a true finding should be studied further. Despite the potential bias using this selected cohort focusing on multiplex families, the CD-associated predominant dysbiosis we found is in agreement with previous findings.
In conclusion, we provide for the first time a set of five bacterial species that characterise the predominant dysbiosis in CD. We identified a faecal microbiota dysbiosis signature associated with CD, characterised by a decreased presence of F prausnitzii, B adolescentis, D invisus and an unknown species of Clostridium cluster XIVa, and an increased presence of R gnavus. The disproportions of three of these predominant species were also quantitatively confirmed, and the identification suggests lack of butyrate-producing capacity in conjunction with mucin degradation as plausible physiopathological explanations. Furthermore, the novelty in the experimental approach is that not only were the predominant microbiota of patients with CD and healthy controls studied, but for the first time comparisons with a large cohort of unaffected relatives of patients with CD were also included. In contrast to CD-associated dysbiosis, the dysbiosis in relatives of patients was not characterised by a lack of butyrate-producing capacity, but similar enhanced mucin degradation could be hypothesised. As the mucosal barrier is the first-line defence of the host against intestinal bacteria, the shift from normobiosis to the dysbiosis observed in relatives of patients with CD might be an intermediate step towards CD and disease-associated dysbiosis. Despite the fact that we did not study the overall butyrate-producing or mucin degradation capacity of the microbiota in this cohort, it is intriguing to find a functional overlap between dysbalanced bacteria in patients with CD and their unaffected relatives at risk. Further investigation should reveal not only if the balance between butyrate production and mucin degradation is indeed essential in predisposing to CD but also how this balance can be influenced.
References
Supplementary materials
online only appendix
Files in this Data Supplement:
Online only appendix
Files in this Data Supplement:
Footnotes
Funding The project was funded by the Fund for Scientific Research-Flanders, Belgium (FWO-Vlaanderen), research project G.0455.06N. GH, SV and VDP are postdoctoral fellows of the FWO-Vlaanderen.
Competing interests None.
Ethics approval This study was conducted with the approval of the ethics committee of the Catholic University of Leuven.
Provenance and peer review Not commissioned; externally peer reviewed.