Article Text

Statistics from Altmetric.com
- Microbiome
- genetic predisposition
- bacteroidetes
- high-throughput DNA sequencing
- Crohn's disease
- inflammatory bowel disease
- intestinal bacteria
- immune response
- mucosal immunology
- bacterial interactions
- metagenomics
- carbohydrates
- anaerobic bacterial fermentation
- molecular biology
- mucins
- intestinal microbiology
- inflammatory diseases
- bacterial adherence
- short-chain fatty acids
- bacterial interactions
- trefoil peptides
- IBD
- mucus
- Helicobacter pylori
- acid-related diseases
- non-ulcer dyspepsia
- genetic polymorphisms
- gastric neoplasia
- colonic microflora
- gene mutation
- IBD–genetics
We read with great interest the recently published study by Rehman and colleagues.1 Investigating microbial diversity on faecal and ileal samples from 48 mice using Sanger and 454 techniques, Rehman et al detected increased percentages of Bacteroidetes in the faeces of adult Nod2−/− mice, whereas Firmicutes levels were significantly higher only in the ileum of Nod2−/− mice.
Since we demonstrated that antibiotic treatment of Nod2−/− mice decreased the number of Peyer's patches and that suppression of the intestinal microbiota was able to fully reverse Nod2−/− mice phenotype,2 we further analysed the faeces of 30 C57BL/6 Nod2−/− (mean age=13 weeks; sex ratio=1) and 30 C57BL/6 wild-type (WT) mice (mean age=10 weeks; sex ratio=1). Age did not differ significantly between mice groups. The V3–V4 region of the bacterial 16S ribosomal DNA was sequenced on a 454 FLX-Titanium pyrosequencer, and 219 964 sequences (average 3660 sequences/sample) passed the quality trimming (size >350 bp/base quality >20). We further used the Ribosomal Database Project Classifier and CD-HIT (Cluster Database at High Identity with Tolerance) Program3 to accurately assign sequences to bacterial genera and down to molecular species (97% similarity clustering).
Our results, in agreement with those by Rehman et al, showed a decreased diversity (Simpson index; WT: 0.03; Nod2−/−: 0.07; p<0.001) and richness (Shannon index; WT: 3.94; Nod2−/−: 3.34; p<0.001) in Nod2−/− mice microbiota. Analysis of phyla distribution highlighted a depletion in bacteria belonging to Proteobacteria (WT: 2.38±1.82%; Nod2−/−: 0.34±0.52%; p<0.001) in Nod2−/− mice microbiota. Top–down analysis of Proteobacteria shed light on two sequence clusters absent from Nod2−/− mice microbiota that related to Helicobacter hepaticus and a Desulfovibrio spp. (figure 1). No significant differences were observed for Bacteroidetes or Firmicutes phyla repartition.
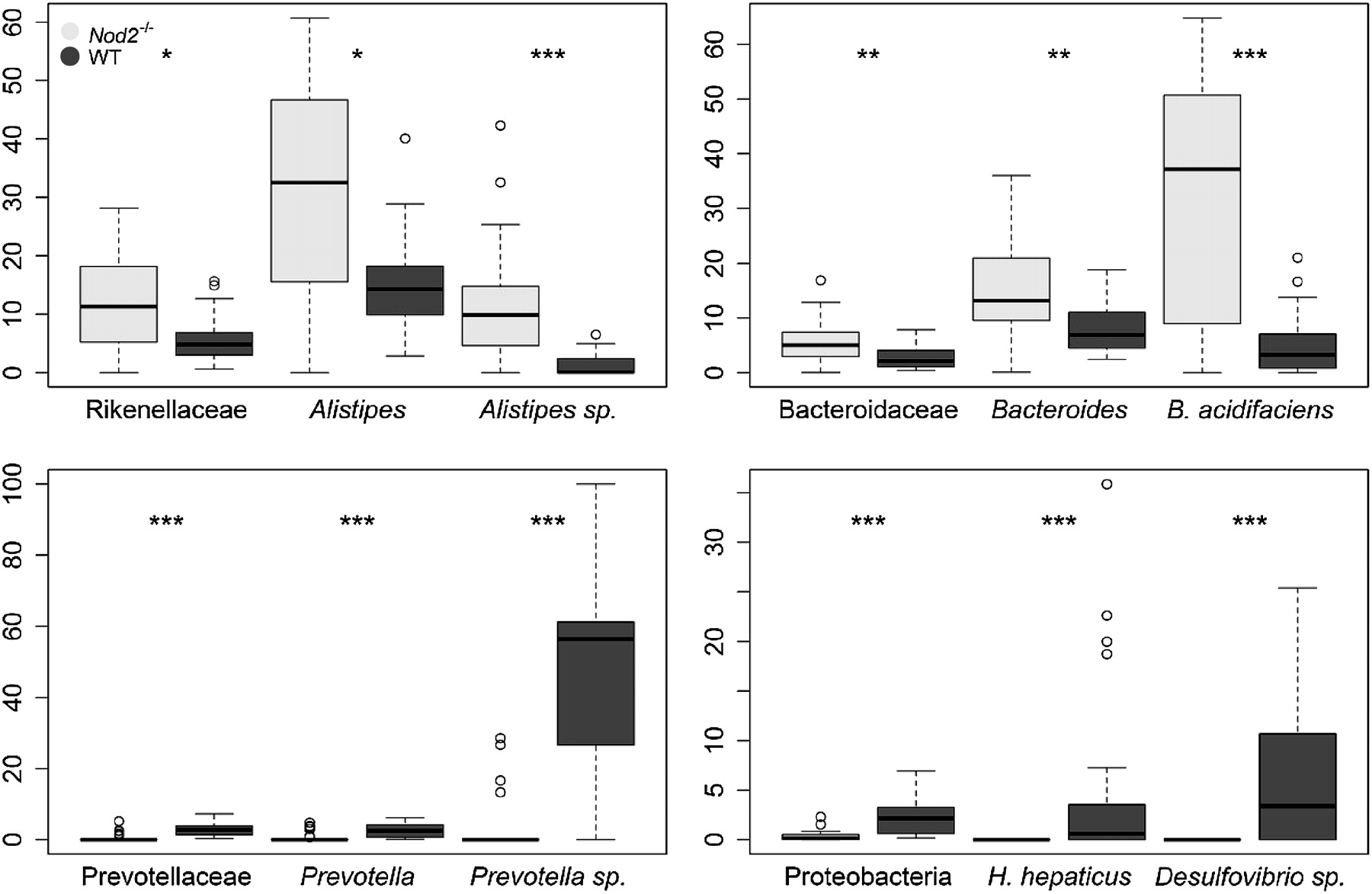
Taxonomic top–down analysis showing significant differences between wild-type (WT) and Nod2−/− mice microbiota. Y-axis represents the percentage of sequences within the upper closest taxonomic level. p Values were computed with a Wilcoxon test and adjusted using Bonferroni correction (*p<0.05; **p<0.01; ***p<0.001). Dark plots represent WT mice microbiota, and light grey plots represent Nod2−/− mice microbiota.
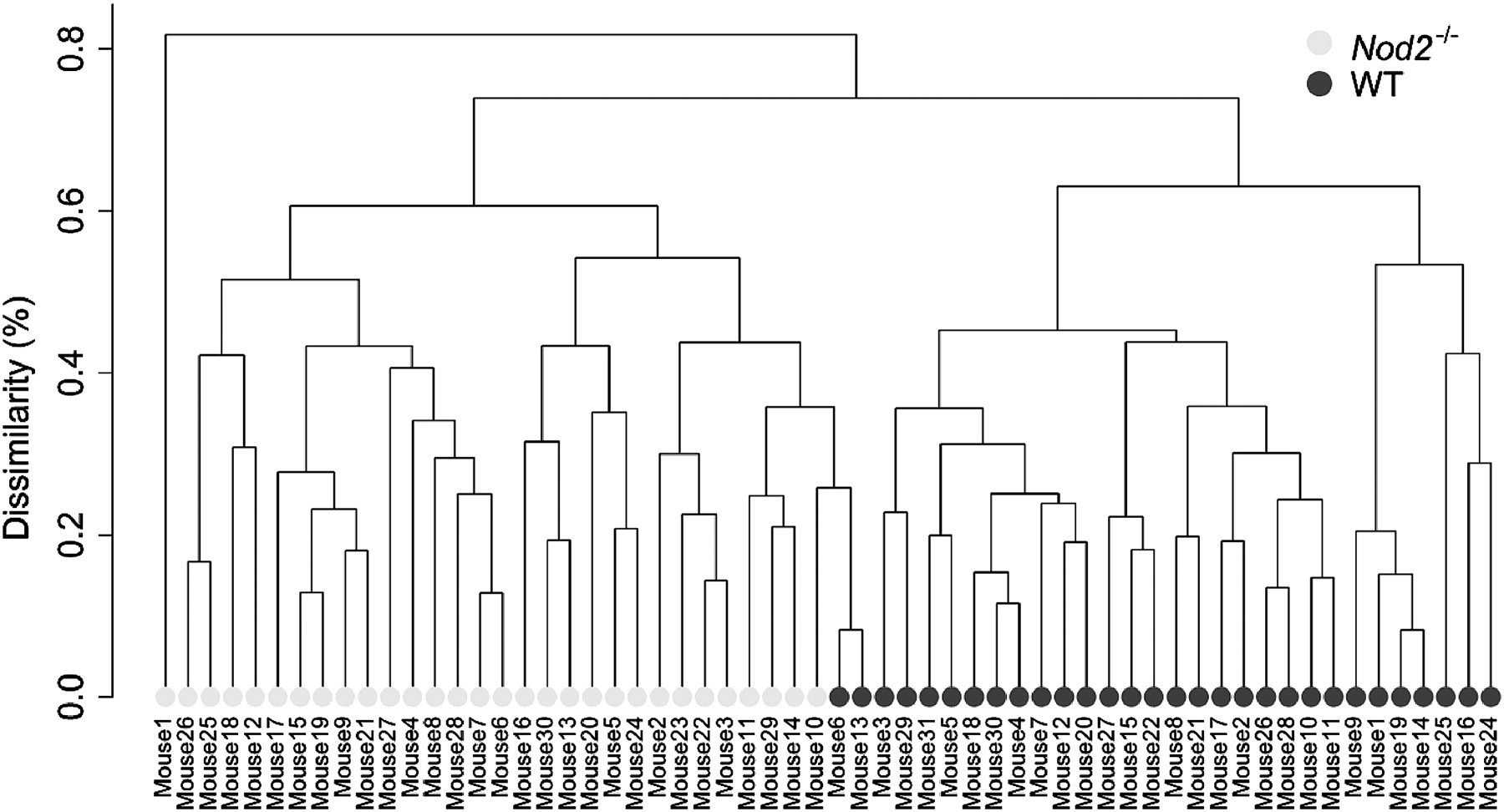
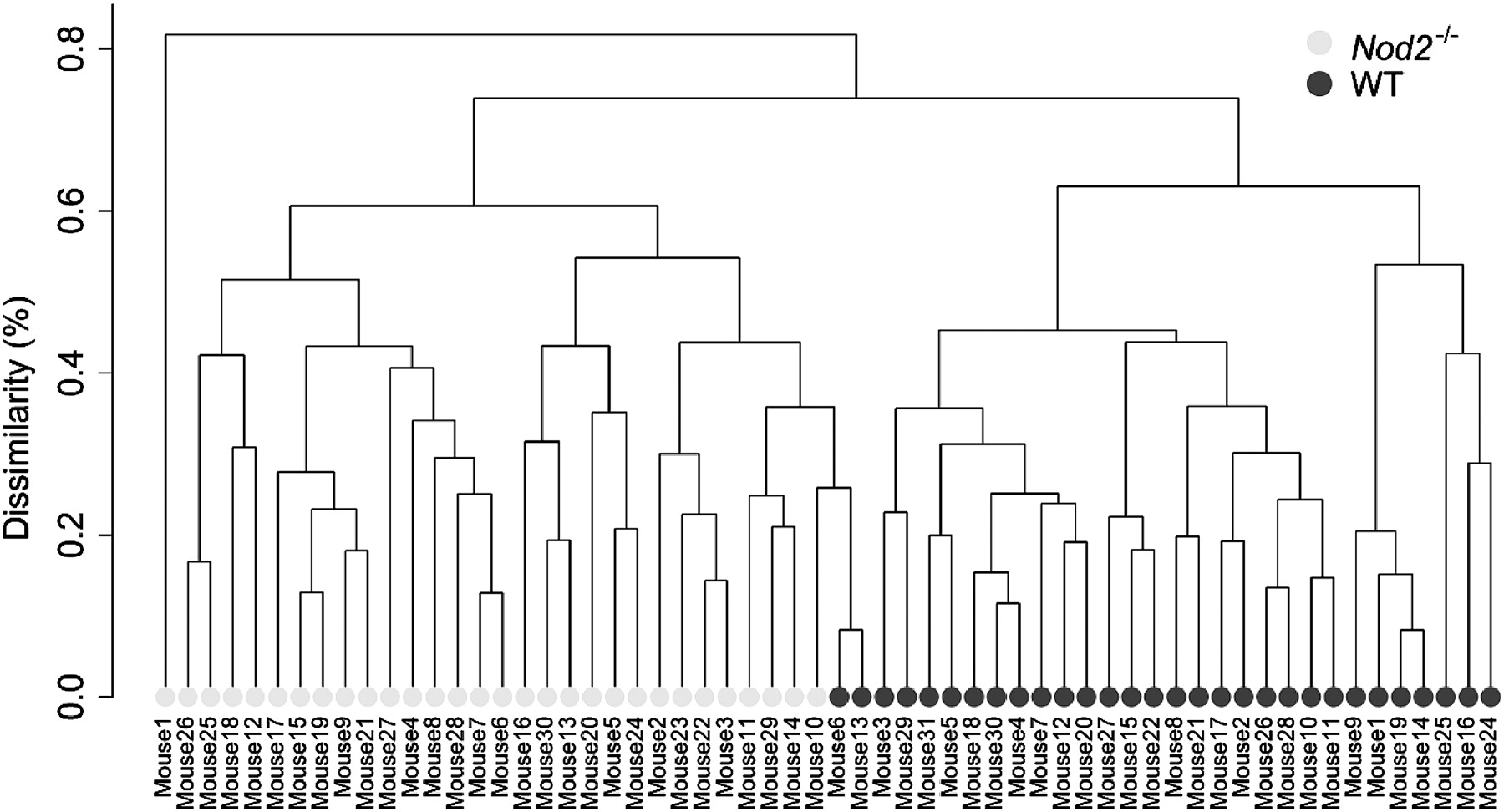
Furthermore, at the genus level, the mice microbiota clustered according to their phenotypes (figure 2), stressing the importance of Nod2 for the control of the intestinal microbiota composition. More specifically, 3 out of the 79 detected bacterial genera were significantly over-represented or under-represented within Nod2−/− mice microbiota. It is noteworthy that these 3 genera were all from the Bacteroidetes phylum. The Alistipes (Rikenellaceae) and Bacteroides (Bacteroidaceae) genera were over-represented in Nod2−/− mice microbiota. Conversely, the Prevotella (Prevotellaceae) genus, with sequences related to an uncultured Prevotella species, was under-represented in these mice (figure 1). Very interestingly, similarly to Rehman et al, we detected Bacteroides acidifaciens as being over-represented in Nod2−/− mice microbiota. This species, adapted to the murine ecosystem and not yet detected in human gut microbiome, is only known to lower pH in vitro and tolerate 20% of bile acid.
{kind=link}
{kind=link}
Nod2 knock out and genus-based microbiota composition. Dendogram drawn using a hierarchical clustering approach on all samples based on bacterial genus distributions. Dark full circles represent wild-type mice (WT) microbiota, and light grey full circles represent Nod2−/− mice microbiota.
Since a dysbiosis within the Bacteroidetes phylum seems confirmed, the interaction of B. acidifaciens with the mice immune system and epithelium permeability deserves further study. A recent study described how Bacteroides fragilis, using an immunomodulatory bacterial molecule (PSA) to shape the host immune responses towards mutualism, secures its persistence on mucosal surfaces.4 One could speculate that the bacterial disequilibrium within a Nod2−/− host microbiome may downregulate these mutualism activities. The altered Nod2−/− mice microbiota also suggests a host failure to regulate the persistence of bacteria belonging to Proteobacteria phylum such as H. hepaticus and Desulfovibrio species. Even though Nod2−/− mice are not diseased, their abnormal microbiota composition combined with deleterious genetic background leads to an impaired gastrointestinal system that may strengthen their predisposition to inflammatory bowel disease.
Footnotes
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.