Article Text

Abstract
Objective Patients with rheumatoid arthritis (RA) have an increased risk of serious infections. Comparing infection rates across RA populations is complicated by differences in background infection risk, population composition and study methodology. We measured infection rates from five RA registries globally, with the aim to contextualise infection rates from an RA clinical trials population.
Methods We used data from Consortium of Rheumatology Research of North America (CORRONA) (USA), Swedish Rheumatology Quality of Care Register (Sweden), Norfolk Arthritis Register (UK), CORRONA International (multiple countries) and Institute of Rheumatology Rheumatoid Arthritis (Japan) and an RA clinical trial programme (fostamatinib). Within each registry, we analysed a main cohort of all patients with RA from January 2000 to last available data. Infection definitions were harmonised across registries. Sensitivity analyses to address potential confounding explored subcohorts defined by disease activity, treatment change and/or prior comorbidities and restriction by calendar time or follow-up. Rates of infections were estimated and standardised to the trial population for age/sex and, in one sensitivity analysis also, for Health Assessment Questionnaire (HAQ) score.
Results Overall, age/sex-standardised rates of hospitalised infection were quite consistent across registries (range 1.14–1.62 per 100 patient-years). Higher and more consistent rates across registries and with the trial programme overall were seen when adding standardisation for HAQ score (registry range 1.86–2.18, trials rate 2.92) or restricting to a treatment initiation subcohort followed for 18 months (registry range 0.99–2.84, trials rate 2.74).
Conclusion This prospective, coordinated analysis of RA registries provided incidence rate estimates for infection events to contextualise infection rates from an RA clinical trial programme and demonstrated relative comparability of hospitalised infection rates across registries.
- rheumatoid arthritis
- epidemiology
- infections
- outcomes research
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Patients with rheumatoid arthritis (RA) have an increased risk of serious infections, and the incidence of infections is affected by many factors.
What does this study add?
We have compared the infection rate in five large registries of RA and one clinical trial programme by harmonising the definition of infection, and we found that, overall, age/sex-standardised rates of hospitalised infection were quite consistent across registries, and with the incidence rate of patients in the fostamatinib clinical trial programme, which was the motivating factor behind this study.
This was especially so when standardising rates additionally for Health Assessment Questionnaire score, a measure of frailty.
How might this impact on clinical practice?
With appropriate standardisation, hospitalised infection rates were reasonably comparable across the RA registries.
Good understanding of underlying infection rates, and determinants for their variation, is important clinically when evaluating potential infection adverse effects of RA treatments, both in clinical practice and in drug development and approval.
Introduction
Patients with rheumatoid arthritis (RA) have an increased risk of infection due to both direct disease-related effects and immunosuppressive treatment-related effects of RA therapies (eg, corticosteroids and tumour necrosis factor antagonists).1–7 For ethical reasons, modern RA drug trials are generally limited to 6 months of placebo-controlled follow-up, and patients without response in any study arm can be rescued to active treatment. Consequently, placebo-arm data are quite limited in both patient numbers and follow-up duration, adding uncertainty around the safety profile of new products for rare and long-term outcomes. Observational data may be used to provide background rates as context for safety events observed in clinical trial programmes.8 9 Typically, published data have been used for such purposes, but reliance on published data has challenging limitations, including differences in patient populations, geographical differences, variability in outcome definitions, lack of concurrent data and analyses that are inadequate for the specific question at hand (eg, typically only a crude overall rate rather than age/sex stratum-specific rates).
We sought to improve on existing methodology for contextualising trial data from the active treatment group with observational data, in order to support safety assessment for an RA drug development programme, given the limited placebo data from the trial programme. By context, it is understood ‘to place (a word, event, etc.) into a particular or appropriate context for the purpose of interpretation or analysis’, that is, here specifically to provide such external context for infection rates observed in the trials. The specific drug, fostamatinib, an oral Syk inhibitor, was being developed for the treatment of RA but was discontinued in this indication following inadequate phase III efficacy results.10–12 While the phase III programme was ongoing, we established a prospective, coordinated approach across multiple RA registries to compile, analyse and interpret real-world safety data in patients with RA to contextualise the clinical trial programme.13–16 Here, we describe and compare real-world rates of infection in patients with RA from diverse regions globally and discuss how these provide context to rates of infection observed in a clinical trial programme.
Methods
The methods of the overall safety contextualisation programme have been described elsewhere.13 In brief, we: (A) included several existing registries with individual-level patient data on infection and established a new registry to enhance geographic coverage across Eastern Europe, Latin America and Asia; (B) harmonised infection outcome definitions across registries; (C) identified baseline differences in demographics, disease history and disease activity between registry cohorts and the clinical trial programme; (D) identified key predictors for infection incidence in the registries; (E) calculated infection incidence rates stratified by these predictors; (F) computed aggregate-level incidences standardised to the clinical trial programme; and (G) assessed their robustness across sensitivity analyses including different definitions of registry cohorts and follow-up times.
RA registries
Five RA registries—Consortium of Rheumatology Research of North America (CORRONA; USA),17–19 Swedish Rheumatology Quality of Care Register (SRR; Sweden),20–22 Norfolk Arthritis Register (NOAR; UK),23–25 CORRONA International (a new multinational registry)13 26 and Institute of Rheumatology Rheumatoid Arthritis Cohort (IORRA; Japan)6 7 27–29—were selected. Selection was based on several considerations, partly focused on optimising comparability with patient populations in a typical RA clinical trial programme: (1) well-established, scientifically rigorous registries with appropriate design and quality for this study; (2) global representation to better geographically match the clinical trial programme; (3) size and data quality, including availability of longitudinal RA-specific data (eg, disease activity measures and treatments); and (4) longitudinal capture of comorbidity events with sufficient data detail and validity.
Features of each registry are described in detail in table 1. CORRONA, established in 2001, is a US-based longitudinal registry of >25 000 patients with RA.17–19 SRR was initiated in the mid-1990s and currently encompasses >40 000 patients with RA.20–22 NOAR, established in 1989, is an early arthritis inception cohort registry that currently comprises >4000 patients, identified from primary and secondary care.23–25 The CORRONA International registry, based on CORRONA US methodology, was set up in 2011 primarily for this study as a stand-alone multinational registry in a collaboration with the study sponsor to enhance geographic representativeness of the registries included in this programme. Information about outcomes in general, and adverse events specifically, were selected and defined influenced by the goals of the overall project ensuring that CORRONA International data collection paralleled the definitions commonly used in clinical trials. The mechanism of ensuring data quality and validity followed the established model of CORRONA US, using automated electronic algorithms to identify missing, erroneous or conflicting entry of data in addition to a thorough data quality control performed by a dedicated CORRONA team.19 By the end of 2012, 5790 patients had been included into this registry from 10 countries in Latin America (Brazil, Mexico and Argentina), Central/Eastern Europe (Czech Republic, Hungary, Poland, Romania, Russia and Ukraine) and Asia (India).13 26 The IORRA cohort was established in 2000 in Japan. Annually, 5000–5800 patients participate, and the total number of patients who have been examined at least once exceeds 11 000.6 7 27–29
Details of the five included real-world RA registries
The clinical trial programme
The clinical trial data included data from all fostamatinib-exposed patients in the phase II-III RA programme including open-label extension studies, with data cut-off of 19 March 2013 (n=3240).10–12 Key relevant exclusion criteria of the overall programme for the current study were recent steroid use, active or recent significant infections and neutropaenia.
Definition of study cohorts and subcohorts
Within each registry, we defined a main cohort of all adult patients (age 18 years or older) diagnosed with RA according to each registry’s definition, from 1 January 2000 or the earliest time point thereafter. No additional inclusion/exclusion criteria were used.
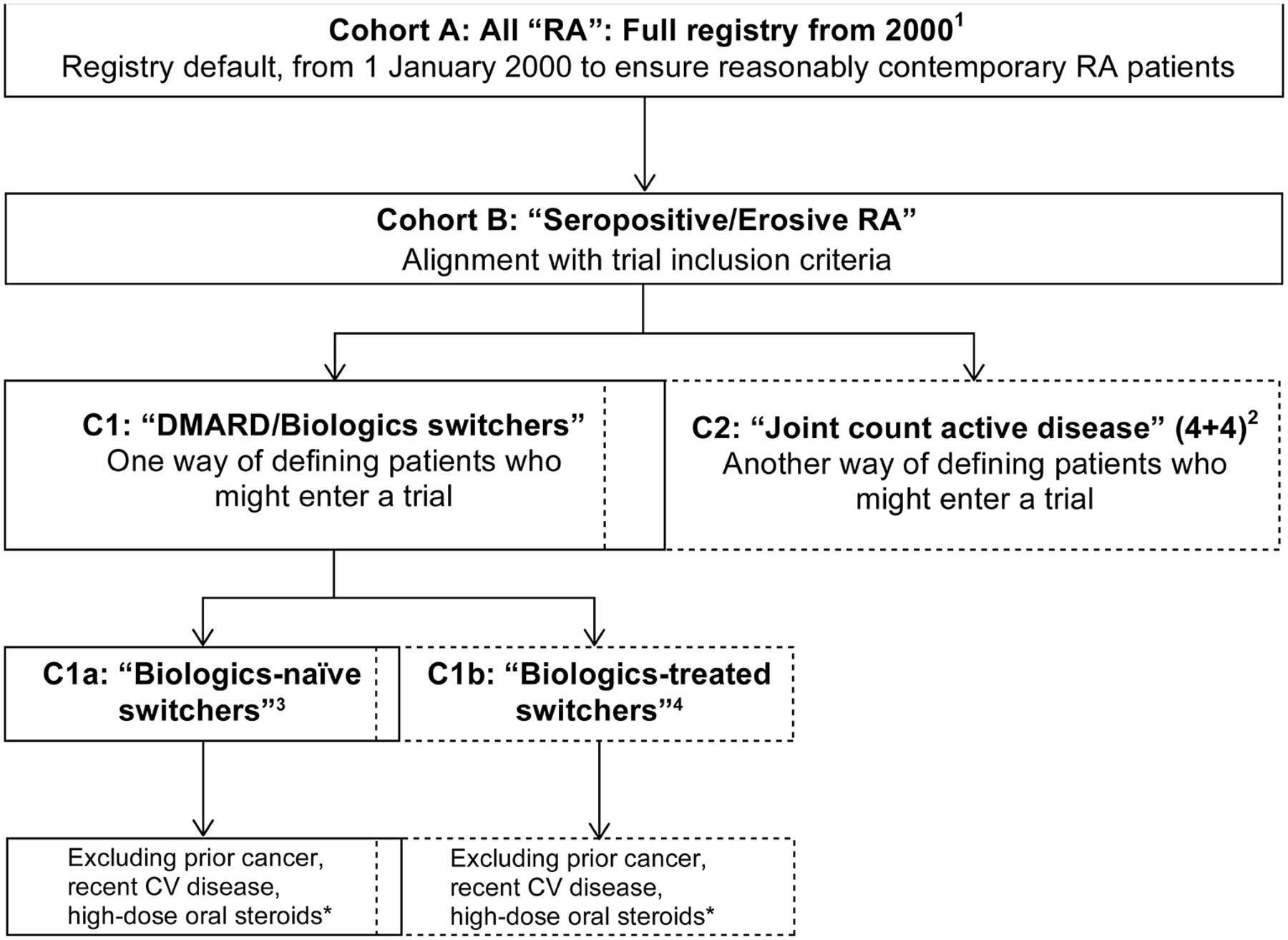
To evaluate the influence of alternative cohort selection criteria on infection incidence and comparability with the clinical trial population, we created subcohorts for sensitivity analysis, defined by criteria related to: disease activity, treatment status/change and/or prior comorbidities (figure 1).
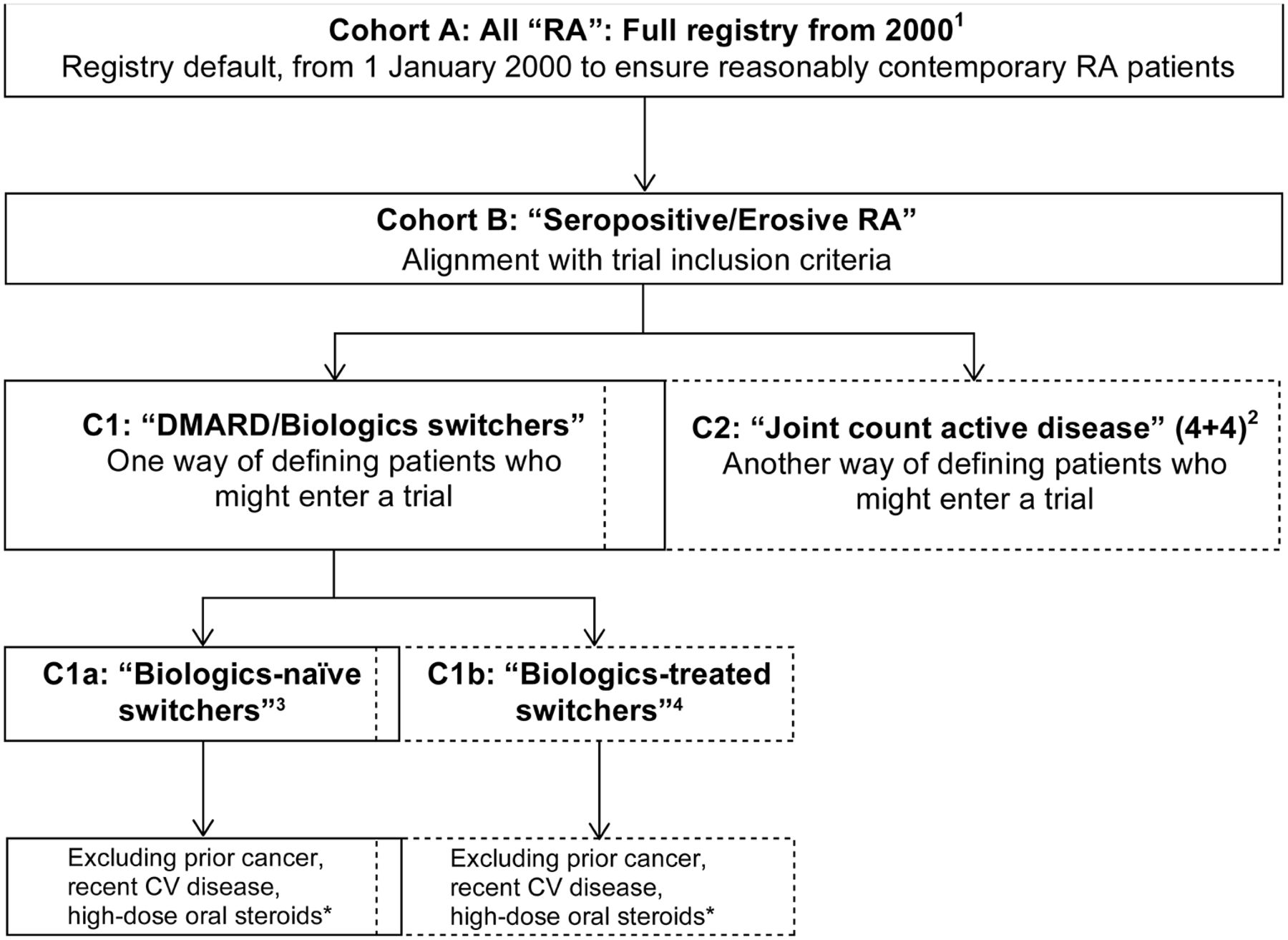
Definitions and relationship between the main cohort (here denoted cohort A) and nested subcohorts within the registries that are used for the main and some subcohort analyses. Note that cohorts B and C1 are intermediate steps and were not included in the final subcohort sensitivity analyses. Note also that the C1 and C2, as well as the C1a and C1b, subcohorts are partly overlapping, as some individuals may be included in one subcohort at one point in time and then at another (later) point in the other (given the longitudinal character of the data). 1Main registry cohort of patients in registry from 1 January 2000 (or earliest first date after 1 January 2000) and baseline at 1 January 2000 or cohort entry. 2Subcohort defined by selection of patients with active RA (patients with seropositive/erosive RA and with >4+4 tender/swollen joints from 28-joint counts). 3Subcohort defined by selection of patients with treatment switch/addition who are previously biologicals naïve (inadequate response to MTX/DMARDs). 4Subcohort defined by selection of patients with treatment switch/addition who have been treated previously with biologicals (inadequate response to biologicals). *At time of treatment change/initiation (‘switch’), excludes patients with history of cancer; major CV event in the previous 6 months (myocardial infarction, unstable angina, stroke, pulmonary embolism or heart failure); oral steroids prednisolone (or equivalent) >10 mg/day—as markers of potential risk for cancer, CV disease and infections—in alignment with the corresponding relevant trial exclusion criteria. CV, cardiovascular; DMARDs, disease-modifying antirheumatic drugs; MTX, methotrexate; RA, rheumatoid arthritis.
Data collection and baseline characteristics
For each analysis, baseline was defined consistently across registries as the point when registry patients fulfilled the cohort membership conditions in each main or sensitivity analysis. Baseline characteristics in each registry were then defined based on available data (online supplementary table 1). In all cohorts, start and stop dates of RA therapies were recorded at each follow-up visit, and most other characteristics were updated regularly depending on data collection and visit schedules, often at each visit, and were used both to define baseline characteristics in each main cohort or subcohort at the respective index date and to define subcohorts as described above. Baseline demographic and disease activity measures were categorised in predefined categories and included age (<50, 50–65 and ≥65 years), gender, 28-joint disease activity score (DAS28; <3.2, 3.2–5.1 and ≥5.1), C reactive protein (CRP) (<6, 6–10, 10–16 and ≥16 mg/L), erythrocyte sedimentation rate (ESR) (<28, 28–42 and ≥42 mm/h), rheumatoid factor (RF) positivity (RF + yes/no) and nationally validated Health Assessment Questionnaire (HAQ) score (<1.1, 1.1–1.8 and ≥1.8).
Supplemental material
Outcome definitions and follow-up
The primary infection outcome was hospitalised infection. In addition, tuberculosis and herpes zoster were also included as specific infection outcomes. Outcome definitions were agreed with each registry to optimise validity and comparability across registries and the clinical trial dataset, depending on the type and level of data available in each registry (online supplementary table 2). Briefly, the definition of hospitalised infection was based on reported infection-related serious adverse events hospitalised for this cause in the clinical trials, linked inpatient registry infection discharge codes in SRR and NOAR, patient-reported infection hospitalisation in IORRA and physician-reported infection hospitalisations in CORRONA and CORRONA International. In each analysis, only the first event of the relevant outcome during follow-up was included, both for trial and registry data.
For the primary analysis, follow-up began at the later date of entry for a patient into the registry or 1 January 2000. In the subcohorts, follow-up began at the later date of entry into the subcohort (based on subcohort eligibility criteria) or 1 January 2000. In all analyses, follow-up ended at the earliest of the following: outcome, death, last date of follow-up in the registry, loss to follow-up or 31 July 2013.
Predictors of infection events in each registry
For each outcome and registry, we assessed the association between predefined covariates and the outcome using Cox regression. The covariates included age, sex, HAQ score, BMI, RA treatment history, indices of baseline RA disease activity and several typical trial exclusion criteria related to comorbidity,10–12 including recent steroid treatment, relevant in this study. These analyses determined which covariates would be used for standardisation of incidences from the RA registries. Apart from age and sex, HAQ score was the strongest predictor of infection events across registries. Consequently, age (four categories) and sex were used as basic standardisation factors (ie, eight strata), and HAQ score (three categories) was added for standardisation in one sensitivity analysis (ie, a total of 24 strata).
Sensitivity analyses
Sensitivity analyses were conducted by changing the variables of the main analysis , and were used to assess the robustness of estimated rates from each registry to confounding by various factors (online supplementary table 3). The sensitivity analyses applied subcohort definitions including selected study inclusion and exclusion criteria to reflect typical trial criteria (figure 1), restricted follow-up time (in calendar time or by truncation at 18 months) and introduced additional standardisation for HAQ score. Sensitivity analyses were only applied to the main outcome (hospitalised infections).
Data management and statistical analysis
Each registry provided counts and percentages of descriptive characteristics at baseline. For the follow-up, they provided number of cases and associated person-time for each study outcome, within strata defined by age, sex and HAQ score. A central analysis used the stratum-specific data to calculate incidence rates for each registry, standardised to the distribution of the RA trial programme patient population for the selected standardisation variables, with CIs based on a gamma distribution.30 31 The clinical trial rate was calculated with exact Poisson 95% CIs. The estimated incidence rates were based on individual records with non-missing observations on all variables involved (outcome, age, sex, and—where used—HAQ score). No patients had missing age or sex, whereas 5.4% of patients had missing HAQ scores.
Individual data were maintained by each registry throughout the study, and all analyses of patient-level data were done locally within the registry for each RA cohort, without pooling of individual data across registries. Study members and registry staff remained blinded to trial data that were not already in the public domain. All registries had appropriate ethical approvals and patient consents.
Results
Baseline characteristics
Table 2 summarises the numbers of patients and the baseline characteristics for each registry’s main cohort and the trial population. Demographic characteristics at baseline of the main registry cohorts showed typical features of patients with RA in a real-world setting, as females account for 70%–85% of the population and 35.4%–54.1% of the patients were 60 years of age or older at baseline; in contrast, the trial population was younger due to the range of trial inclusion and exclusion criteria applied but with a similar sex distribution. There were larger differences in baseline disease activity and clinical characteristics. Patients from the trial programme had higher DAS28, HAQ scores, swollen joint counts (data not shown) and ESR values than registry patients, whereas differences in CRP were less pronounced (table 2). There were also differences between the trial population and the registry cohorts and across these cohorts, with regards to baseline comorbidities (with the definitions available for each data source) (table 2). Treatment from baseline varied considerably across registries and the trial population, reflecting the varying selection criteria, as well as background differences in the various populations. In the different sensitivity analyses, some of these baseline differences were reduced; for example, in sensitivity analyses 2 and 7, the biologicals-naïve patients switching or adding treatment (inadequate responders to methotrexate/disease-modifying antirheumatic drug (MTX/DMARD-IR)), MTX treatment was more similar across registries, although many differences remained (online supplementary table 4).
Baseline characteristics of patients with RA in the main cohorts from five RA registries and one RA clinical trial programme
Incidence of hospitalised infections, tuberculosis and herpes zoster
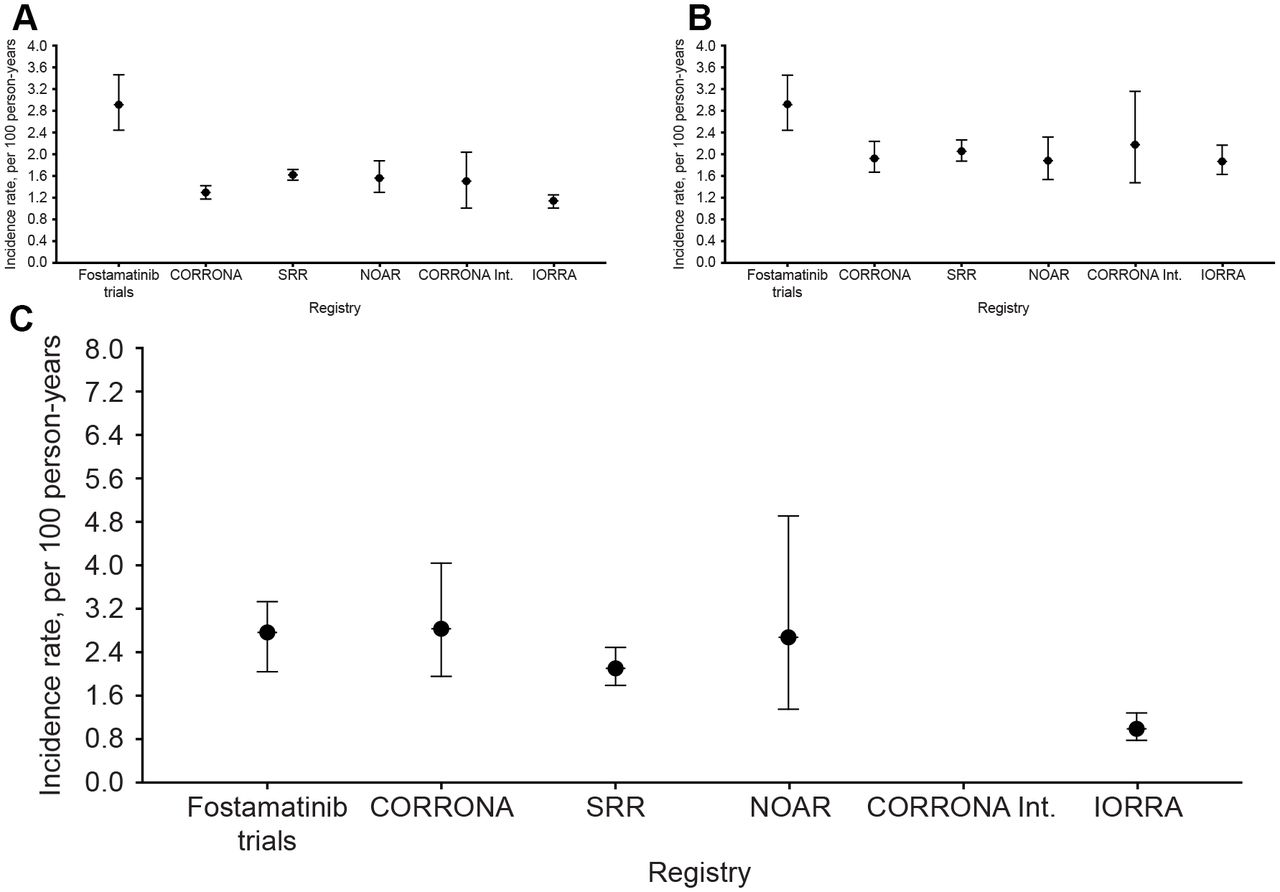
In the main analysis, crude rates of hospitalised infections differed quite notably across the five registry cohorts (range 1.16–2.48 per 100 person-years (PY)), whereas after age-standardised and sex-standardised incidence rates were fairly consistent across the registry cohorts (range 1.14–1.62 per 100 PY). The rate was higher in the RA trial programme cohort (2.92 per 100 PY) (tables 3 and 4, figure 2A).
Numbers of events (person-time) and incidence rates of infections per 100 PY standardised by age and sex* (with 95% CIs)†, in the main cohorts from five RA registries and one RA clinical trial programme

{kind=link}
{kind=link}
Incidence rates of hospitalised infection per 100 person-years, standardised for age and sex, among patients with RA from five RA registries and one RA clinical trial programme in the main analysis and two sensitivity analyses: (A) main analysis (main cohort); (B) main cohort, standardised for HAQ score in addition to sex and age and (C) restricted to biologicals-naïve (MTX/DMARD-IR) patients, with 18 months truncated follow-up. Approximate CIs based on the gamma distribution for registry data and exact Poisson CIs for fostamatinib data. For panels (A) and (C), incidence rates for the registry data were standardised according to the age and sex distribution in the appropriate corresponding cohort (A: overall cohort, C: MTX-IR cohort) of the fostamatinib dataset; for fostamatinib, crude incidence is presented. For panel (B), incidence rates for the registry data were standardised according to the age, sex and HAQ score distribution in the overall cohort of the fostamatinib dataset; for fostamatinib, crude incidence is presented. CORRONA, Consortium of Rheumatology Research of North America; HAQ, Health Assessment Questionnaire; IORRA, Institute of Rheumatology Rheumatoid Arthritis; MTX/DMARD-IR, Methotrexate/Disease Modifying Anti Rheumatic Drug-Incomplete Response; NOAR, Norfolk Arthritis Register; RA, rheumatoid arthritis; SRR, Swedish Rheumatology Quality of Care Register;
Hospitalised infection incidence rates per 100 person-years standardised by age and sex, in data from five RA registries and one RA clinical trial programme, across all sensitivity analyses.
For a number of the sensitivity analyses, the standardised incidence rates of hospitalised infection showed a higher consistency across the registry subcohorts and with the trial data. This was particularly evident for the analysis standardised for HAQ score in addition to sex and age (sensitivity analysis 9), where the registry rates were higher than in each of the main registry cohorts (range 1.86–2.18 per 100 PY across registry subcohorts), and more consistent with the incidence rate 2.92 per 100 PY in the full RA trial cohort (table 4, figure 2B). A similar tendency to greater similarity across datasets (except IORRA) was seen for sensitivity analysis 7 in biologicals-naïve patients switching or adding treatment (MTX/DMARD-IR), focusing on the first 18 months after treatment initiation (table 4, figure 2C; range 2.10–2.84 per 100 PY across registry subcohorts except IORRA), where the rate in the RA trial programme MTX-IR cohort was 2.74 per 100 PY.
Incidence of tuberculosis and herpes zoster was low in all cohorts, and in some cases so rare that stable rates could not be estimated. Standardised incidence rates for tuberculosis in the main cohorts were lower in CORRONA and SRR compared with IORRA and CORRONA International, largely reflecting known regional differences (table 3). Similarly, standardised incidence rates for hospitalised herpes zoster and all herpes zoster were lower in CORRONA and CORRONA International compared with IORRA (table 3). In the trial programme, exclusion criteria for latent tuberculosis infection should reduce the incidence; although events were few, rates appeared to be within the range of the registries for these outcomes.
Discussion
In this study, we have successfully implemented a novel approach to using observational data for contextualising infection rates observed in a clinical trial. Through a prospective, coordinated analysis of several RA registries, we have demonstrated the ability to provide improved consistency of data across registries, resulting in more extensive and comparable information for contextualising trial rates.
It is interesting to note that incidence rates of hospitalised infections, which as crude measures differed quite notably across five registry cohorts (range 1.16–2.48 per 100 PY, ie, a ratio of 2.1), were considerably more consistent when standardised for age and sex (range 1.14–1.62 per 100 PY, ratio 1.4). This indicates that age/gender standardisation to reduce confounding is critical for comparing infection rates across registries and when using them to contextualise a global clinical trial programme. Further standardisation for HAQ score (beyond just age and sex) also appeared to be of importance to provide more comparable data from the registries, providing higher standardised rates for most registries (reflective of the higher HAQ scores among trial participants) and closer to the trial programme rates. This result is consistent with the importance of general disability in the susceptibility of patients to infections.32 33 Despite differences across registries in, for example, comorbidity and oral corticosteroid treatment, we did not find that these factors were important predictors across registries of infection incidence rates when age, sex and HAQ were already considered. This may relate to differences in the way these are captured in the various registries (which could exaggerate differences) or it may indicate that age, sex and HAQ differences, which better capture infection risk across registries, may be partially correlated with these factors but more consistently measured.
When compared with the RA clinical trial population, registry rates of hospitalised infection were in general lower; however, when sensitivity analyses were conducted, including of subcohorts of patients more similar to those in the trial population, the rates became more similar, suggesting an important influence and potential confounding of these variables, which could be removed by restriction. This implies that despite the fact that many baseline differences appeared to remain also in the sensitivity analyses (eg, online supplementary table 4), any remaining variability in baseline differences across cohorts has limited influence on rates of infection in these analyses. Additionally, for most registries, the rate of infection was higher in the first 18 months following a treatment change, a follow-up duration that is more similar to the trial programme. This finding is consistent with other published results and may reflect an increase in the risk of hospitalised infection related to the period following switch/addition of treatments in patients with active RA, which could be due to true increased risk to susceptible patients during that period and/or more active medical surveillance or management during that time and/or treatment discontinuation by susceptible individuals over time after a treatment switch, as suggested by previous literature.2 5 33 34 Another potential confounder is the availability of reimbursement cover for hospitalisation in a trial setting, which may contribute to higher rates of hospitalisation for trial participants in some countries.
These data therefore suggest that the main analysis in the registries, for example, using all data from 2000 through end of study period, may provide a good estimate of risk in the longer term but may underestimate the short-term risk in the period after a change in treatment (eg, add-on or switch). The sensitivity analysis truncating follow-up after 18 months likely provides a better estimate of the short-term risk of infection in selected populations and may provide a more appropriate context for clinical trial data regarding infection outcomes, given the relatively short follow-up in most clinical trial programmes. The use of consistent baseline confounders, selection of patients meeting trial eligibility and length of follow-up are the key factors to providing contextual rates of hospitalised infection. The adjustments/standardisations are not specific to registry-specific confounders or treatment patterns and illustrate that variability remaining is relatively limited and thus provides interpretation that is not registry specific.
The prospective approach to generating contextual observational data has several strengths that improve on reliance on available published data. In this study, these included: (1) working directly with registries to allow direct access to data sources, enabling appropriately tailored analyses including standardisation and stratification for relevant variables and providing better support for sensitivity analyses and data interpretation; (2) a main analysis using a relatively unselected cohort over long follow-up from each registry (to maximise precision of event rate estimates), supplemented with a series of sensitivity analyses (to assess potential bias and variability in main analysis); (3) the selection of registry subcohorts in some sensitivity analyses to more closely resemble clinical trial populations in order to understand how important these aspects are; (4) temporal matching to the current clinical trial programme to address potential changes in risk panorama, treatment patterns and disease risk over time; and (5) appropriate selection and improved comparability of outcome definitions across registries and with trial data, achieved by tailored registry definitions to better match the definitions obtainable from the clinical trials, as well as from other registries.
Despite our efforts to address and minimise limitations inherent in using observational data to contextualise clinical trial data, some important limitations remain and must be considered when interpreting our results. These include: (1) registry-based cohorts may under-report outcomes if patients leave the registry before events occur or are not adequately followed-up.35 36 (2) Data collection methods for events vary between the clinical studies and the registries. In the clinical programme, events are actively collected by physicians and often adjudicated; in the registries outcome capture differs and can be collected actively or through other systems (eg, linkages to external data sources). Despite these differences, the outcomes assessed here were defined and selected based on feasibility to assess with reasonable validity in both registries and clinical trial data, as well as being of clinical relevance. (3) Clinical trial programmes extensively use inclusion/exclusion criteria and therefore have more homogenous and selected populations compared with typical populations found in registries, which are typically more representative of the general patient population with RA. A clinical trial programme may also be enriched for aspects of the studied disease, for example, higher disease activity in RA.9 28 If these factors affect the outcome studied, bias may ensue. (4) All registries were established independently at different times without initial harmonisation of data definitions. Baseline and variable definitions were harmonised as much as feasible before analysing the data, but remaining variability across registries remains a potential limitation. (5) The influence of potential time-varying confounders, such as changes in treatment for RA, was not part of the predefined analysis plan, and the analyses were therefore limited to evaluating the influence of baseline covariates on infection rates. However, this approach is consistent with the typical methodology used in the analysis of clinical trials, and as the ultimate goal of this work was to contextualise a clinical trial programme with registry data, evaluating baseline covariates as predictors of infection in these cohorts is appropriate. While the extent of either of these potential limitations cannot be fully known, the current study was designed to address them with a variety of sensitivity analyses, and the overall consistency of results across the analyses lends additional weight to the overall findings. In addition, some of the sensitivity analyses provided insights consistent with prior literature of potential factors affecting variation in infection rates.
In conclusion, a coordinated analysis across multiple large RA registries provided stable background incidence rate estimates for infection events, and these estimates were valuable for the contextualisation of an RA clinical trial programme with limited placebo follow-up. This analysis demonstrates unique considerations needed when comparing infectious events between clinical trial and observational data and the need to consider creating more comparable cohorts and analyses for the most appropriate comparisons and contextualisation. Overall, our results provide a considerable improvement on reliance on published studies alone, an attractive approach to contextualising safety data from clinical trials.
References
Footnotes
Contributors All authors were involved in the conception and design of the study and/or analyses. All authors were involved in the data analysis of each cohort and were involved in data interpretation. Drafting of this paper is mainly by HY and FN, but all authors were involved in critically reviewing the manuscript, and all authors read and approved the final version.
Funding AstraZeneca funded this study and collaborated with the researchers in the design, analysis and interpretation. Five independent registries have own funding support, but those funding sources are not involved in the design, analysis of this study. NOAR is funded by Arthritis Research UK. SRR has or has had research agreements with AbbVie, Pfizer, BMS, UCB, Merck, AstraZeneca, Sobi, and Roche. IORRA is supported by various grants from a large number of pharmaceutical companies, including AstraZeneca. CORRONA Inc. has received funding in the past 2 years from AbbVie, Amgen, AstraZeneca, Janssen, Genentech, Lilly, Novartis, Pfizer, Regeneron, Vertex and UCB, through contracted subscriptions to the database, and CORRONA International LLC has received funding from AstraZeneca.
Competing interests FN, NB, TNT, EW-vS and MeH are employees of AstraZeneca. FN, NB, LH, TNT, EW-vS, MeH and SF hold AstraZeneca stocks and/or options. LH and SF were employees of AstraZeneca during the time in which the research was conducted, and CG was affiliated with the Arthritis Research UK Centre for Epidemiology. JA has had grant/research support from AstraZeneca and has been a consultant for AstraZeneca. TF has received honoraria for advisory board participation from Pfizer. JDG is a shareholder of CORRONA, Inc., and has consulted for AstraZeneca, CORRONA, Novartis and Pfizer. DAP is an employee of CORRONA and has been a paid instructor for Novartis. GR is an employee of CORRONA. KM has grant/research support from ACR Rheumatology Research Foundation and is codirector of the National Data Bank for Rheumatic Diseases (NDB), which has received funds from AstraZeneca. HY leads the IORRA cohort and has been a consultant for AstraZeneca. DS has had grant/research support from AstraZeneca and has been a consultant for AstraZeneca.
Ethics approval Ethics committee/institutional review board of each institution.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement None.