Article Text

Abstract
Objective To characterise changes in selected haematological parameters following once-daily oral baricitinib dosing.
Methods Data were pooled from eight randomised clinical trials (four phase 3, three phase 2, one phase 1b) and one long-term extension. Changes in haematological parameters were evaluated up to 128 weeks (N=2387); overall safety of baricitinib was assessed up to 6 years (N=3492).
Results Mean absolute neutrophil counts decreased (−1.36×109/L) within 1 month, followed by stabilisation within the normal reference range through week 128. The incidence of serious infections was not elevated in patients with neutropenia during the 24-week placebo-controlled period. Mean lymphocyte counts increased (+0.30×109/L) within 1 month, then decreased to baseline (weeks 12–24). Mean platelet counts increased at week 2 (+51×109/L), then decreased towards baseline. Overall, mean haemoglobin concentrations decreased (−0.12 mmol/L), then returned to baseline; however, reduced baseline haemoglobin concentrations observed in the highest baseline high-sensitivity C reactive protein quartile increased over time. Permanent drug discontinuation occurred due to laboratory abnormalities related to neutrophil count in 8 (0.2%), lymphocyte counts in 6 (0.2%), platelet counts in 8 (0.2%), and haemoglobin levels in 16 (0.5%) of all baricitinib-treated patients (N=3492 with 7993 total person-years of exposure).
Conclusions Moderate decreases in neutrophils were seen during baricitinib treatment; however, serious infection was uncommon in patients with neutropenia. Transient increases were observed in lymphocytes and platelets, which returned to baseline over time. Changes in haemoglobin concentration were generally small. Haematological abnormalities seldom led to drug discontinuation.
- Anti-Inflammatory Agents
- Non-Steroidal
- Antirheumatic Agents
- Arthritis
- Rheumatoid
- Autoimmune Diseases
- Inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Anti-Inflammatory Agents
- Non-Steroidal
- Antirheumatic Agents
- Arthritis
- Rheumatoid
- Autoimmune Diseases
- Inflammation
Key messages
What is already known about this subject?
Baricitinib inhibits JAK2-mediated signal transduction, including that through the erythropoietin receptor which mediates erythrocyte production.
What does this study add?
This study reports changes in haematological parameters that occurred during the first 19 months of treatment across all studies of baricitinib conducted in patients with rheumatoid arthritis.
Baricitinib treatment was associated with decreases in neutrophils that were not associated with severe infections and with transient increases in lymphocytes and platelets, which usually returned to baseline levels and remained stable up to 128 weeks of treatment.
Changes in haemoglobin were generally small-to-moderate, and mostly reversible. Haematological abnormalities infrequently resulted in permanent discontinuation of therapy.
How might this impact on clinical practice?
Characterisation of the impact of baricitinib treatment on haematological parameters contributes to understanding the relative risk and benefits of its use in patients with rheumatoid arthritis.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterised by synovitis, progressive damage to appendicular joints and variable extra-articular manifestations. Medications used to treat RA include conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), biological DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs). However, in clinical practice, despite long-term treatment with combinations of csDMARDs and bDMARDs, some patients with RA do not achieve low disease activity or remission.1
An increased incidence of haematological abnormalities, such as neutropenia, lymphopenia, thrombocytopenia and anaemia, has been observed in patients with RA.2–6 Many drugs used to treat RA may themselves be associated with haematological abnormalities, through differing mechanisms depending upon the drug class.7–11 Additionally, reduction of inflammation with drug treatment has been associated with improvement in anaemia and other haematological abnormalities.11–14
Janus kinase (JAK) inhibitors target cytokine signalling pathways implicated in RA pathogenesis.15 16 In phase 3 clinical trials, treatment with baricitinib, an oral JAK1/JAK2 inhibitor, has improved signs and symptoms of RA, including preventing joint damage and preserving physical function.17–20 Because baricitinib inhibits JAK2-mediated signalling and erythropoietin stimulates erythrocyte production through JAK2 signalling, haemoglobin and other haematological parameters were of particular interest during the clinical development of baricitinib. Here we report changes in key haematological parameters that were observed during clinical trials of baricitinib in patients with RA and discuss their clinical relevance.
METHODS
Study design and patients
Data were pooled from eight randomised clinical trials (four phase 3; three phase 2; one phase 1b) and one ongoing 104-week long-term extension (LTE). Studies included in one or more analyses are described in table 1. Patients were aged ≥18 years and had moderate-to-severely active RA. Key exclusion criteria included haemoglobin <10.0 g/dL (6.1 mmol/L); absolute neutrophil count (ANC) <1.2×109/L; lymphocyte count <0.75×109/L; and platelet count <100×109/L. Study drug was discontinued temporarily if a patient’s laboratory results included ANC <1.0×109/L; lymphocyte count <0.5×109/L; platelet count <75×109/L; or haemoglobin <8.0 g/dL.
Summary of studies
Statistical analyses
A description of the data sets and listing of the analyses in which they were used is presented in table 2. While collection of data for most endpoints (adverse events (AEs), haematological laboratory values, shifts in categorical classification of haematological laboratory values, and temporary and permanent discontinuation due to laboratory abnormalities) was prespecified in each protocol, combined and subgroup analyses were performed on a post hoc basis. The analyses of erythropoietin, reticulocyte count, red blood cell count, and total iron and TIBC in the JADN study were prespecified exploratory analyses.
Data set definitions and list of analyses
Supplemental material
Baseline data were summarised as mean (SD) for continuous variables and n (%) for categorical variables. Laboratory values over time (neutrophils, lymphocytes and platelets) were shown as mean and SD. Changes from baseline over time (haemoglobin) were shown as mean and SE. All analyses used data as observed (no imputation of missing data) with the exception of the analysis of mean neutrophil count by change in high-sensitivity C reactive protein (hsCRP) categories (online supplemental figure 1), which used modified last observation carried forward imputation. For change in mean platelet count and mean platelet volume (MPV) (online supplemental figure 3), data are shown for patients receiving baricitinib 4 mg with baseline and on-treatment count or volume data >0 at a given time point, censored at rescue.

Neutrophils (A) and lymphocytes (C) over time (absolute values) and reversibility assessment for neutrophils (B) and lymphocytes (D).

Mean platelets over time (absolute values) (A) and reversibility assessment for platelets (B).
Assessments
Neutrophil counts, lymphocyte counts, haemoglobin and platelet counts were assessed in blood samples obtained at baseline, during randomised treatment up to 24 weeks, and throughout the LTE. During placebo-controlled, randomised treatment, samples were collected at study weeks 2, 4, 8, 12, 14 (except in the JADA study), 16, 20, and 24. Sampling at later time points differed between studies; for data points including data obtained at different specified study weeks, the range of weeks during which the samples were collected is indicated on the horizontal axis (eg, 48–52 weeks). Neutrophil and lymphocyte counts were categorised by Common Terminology Criteria for Adverse Events (CTCAE) Grade.24 MedDRA-preferred terms for AEs counted as related to particular cell types or haemoglobin were as follows: for neutrophils, ‘neutropenia’, ‘neutrophil count decreased’, ‘febrile neutropenia’ and ‘neutrophilia’; for lymphocytes, ‘lymphopenia’ and ‘lymphocyte decreased’; for platelets, ‘thrombocytosis’, ‘thrombocytopenia’ and ‘platelet count decreased’; for haemoglobin, ‘anaemia’ and ‘haemoglobin decreased’.
Haemoglobin concentrations were categorised as treatment-emergent abnormally low and/or below the gender-adjusted lower limit of normal (gaLLN) based on the following cut-offs: <7.2 mmol/L (11.6 g/dL) for women <59 years and <7.14 mmol/L (11.5 g/dL) for women ≥59 years; <7.88 mmol/L (12.7 g/dL) for men <59 years and <7.76 mmol/L (12.5 g/dL) for men ≥59 years. Treatment-emergent abnormally low haemoglobin was also separately characterised as shifts from baseline CTCAE Grade <2 to post-baseline Grade ≥2 and from baseline Grade <3 to post-baseline Grade ≥3.24
Reversibility was evaluated in a subgroup of patients (see table 2) who discontinued treatment by week 24 and had data available from baseline, last visit and post-treatment assessments. For the analysis of platelet count shifts on treatment (online supplemental table 2), denominators were the number of patients at risk for the shift in each treatment group, which could vary since not all analytes or the same methods were used in every study. Anaemia status at baseline by background medications was assessed in the combined randomised populations of studies JADV and JADX, which both required that patients had previously received csDMARDs or methotrexate (MTX) (see table 1).
Safety assessments included reports of potentially relevant treatment-emergent AEs, serious AEs, and temporary and permanent treatment discontinuations.
RESULTS
Patients
Data were analysed from 3492 treated patients with up to 7993 patient-years of exposure as of 1 April 2017. Patient demographics, including age, duration of RA, and corticosteroid and MTX use, were similar across treatment groups in each analysis set (table 3). The mean ages across the primary randomised treatment data sets for the placebo, baricitinib 2 mg and baricitinib 4 mg treatment arms ranged from 52.9 years to 53.7 years, and was 52.9 years in the all Bari-RA set. In the same data sets, the mean duration of RA since diagnosis was 8.9–9.0 years for the randomised dose groups and was 7.7 years in the all Bari-RA set. The majority of patients in each analysis set were female.
Baseline characteristics
Neutrophils
As shown in figure 1A, baricitinib treatment was associated with an initial decrease in mean neutrophil count at week 4 (−1.36×109/L below baseline) that stabilised by week 12 (−0.87 ×109/L below baseline) and remained stable within the normal reference range through week 128. In an assessment of the reversibility of changes in neutrophils among all evaluable patients who discontinued treatment by week 24, the neutrophil count returned to near baseline levels at the post-treatment visit (figure 1B).
Laboratory abnormality AEs related to neutrophil counts were reported in 60/3492 patients (1.7%) over 7860 patient-years of exposure. Neutropenia was mostly of grade 1, occurred infrequently, and an elevated rate of serious infections was not observed in patients with neutropenia relative to other patients (table 4).
Infection by worst CTCAE grade during placebo-controlled period up to week 24
Temporary interruption of study drug administration due to any neutrophil-related or other haematological laboratory abnormality occurred infrequently (<1%) during the randomised treatment phase (table 5). Neutrophil count–related laboratory abnormality AEs did not lead to permanent discontinuation of study drug during the 24-week placebo-controlled treatment period for any patients in the placebo or baricitinib 2 mg or 4 mg treatment groups. Permanent discontinuation due to neutrophil count-related laboratory abnormalities was reported for 8/3492 patients (0.2%) in the all Bari-RA set (table 5).
Haematological laboratory abnormality adverse events leading to study drug interruption or permanent discontinuation (on-treatment analysis)
To assess the impact of changes in inflammation on neutrophil counts, neutrophil counts were summarised for subgroups of patients with a ≥70% reduction in hsCRP versus those with an increase or a ≤15% reduction in hsCRP through 12 weeks of treatment with baricitinib 4 mg in the RA-BEAM study (online supplemental figure 1). The mean neutrophil count at week 12 for the ≥70% hsCRP reduction subgroup (289/483 patients) was lower by week 8 than that in patients with an increase or a ≤15% reduction (78/483), suggesting an association between greater improvement in inflammation and the magnitude of decrease in neutrophils.25
Lymphocytes
Mean lymphocyte counts increased (+0.30×109/L above baseline) during the first month of baricitinib administration, decreased to baseline levels between weeks 12 and 24 and then remained stable and within the normal reference range through week 128 (figure 1C). In patients assessed for reversibility of changes after discontinuation through 24 weeks, lymphocyte counts returned to baseline at the post-treatment visit (figure 1D). Laboratory abnormalities related to lymphocyte counts were reported in 133/3492 (3.8%) patients. Grade 1 lymphopenia was associated with a slightly elevated rate of overall infections in the baricitinib 4 mg group compared to placebo (81 patients (39.5%) vs 65 patients (27.9%), respectively; table 4). Among all patients with Grade 1 or higher lymphopenia, 6/284 patients (2.1%) treated with baricitinib 4 mg and 4/349 patients (1.1%) who received placebo developed a serious infection.
Lymphocyte count–related laboratory abnormality AEs led to permanent discontinuation during the placebo controlled treatment period for one patient each in the placebo (0.1%) and baricitinib 2 mg (0.2%) and baricitinib 4 mg (0.1%) treatment groups. Permanent discontinuation due to lymphocyte count–related laboratory abnormalities was reported for 6/3492 patients (0.2%) in the all Bari-RA set (table 5).
Platelets
With baricitinib treatment, mean platelet counts increased and peaked at week 2 (+51×109/L compared to baseline), returned towards baseline, and remained stable through week 128 (figure 2). In the subset of patients who were assessed for reversibility of changes through 24 weeks, mean platelet counts were somewhat elevated at the last visit on treatment but returned towards baseline after treatment discontinuation (baricitinib 2 mg: +27×109/L; baricitinib 4 mg: +7×109/L above baseline).
{kind=link}
{kind=link}
{kind=link}
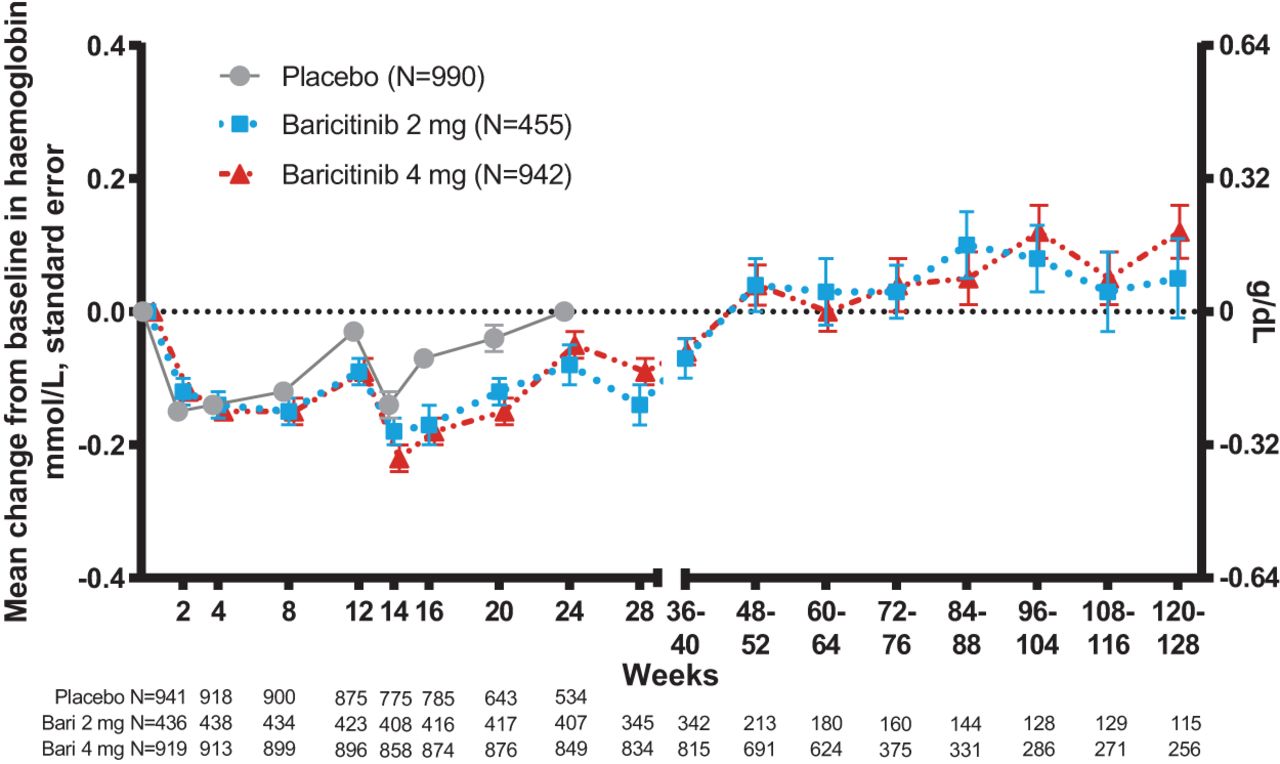
Mean change from baseline in haemoglobin, weeks 0 to 128.
High platelet counts (>ULN; 394×109/L for age ≥60 years, >400×109/L for age <60 years) were found at any time during treatment for 16.9% of patients receiving baricitinib 2 mg and 24.9% of those receiving baricitinib 4 mg versus 9.5% of those receiving placebo; very high (≥600×109/L) levels were found for 1.5% and 2.3% versus 1.3%, respectively (online supplemental table 1).
Changes in platelet counts were also assessed by baseline platelet quartile. Following a transient peak at 2 weeks, platelet counts in the highest baseline quartile decreased slightly and remained below baseline (baseline mean=411.5×109/L); in the two middle quartiles, platelet counts remained stable slightly above baseline; and in the lowest baseline platelet quartile, platelet counts remained above baseline levels throughout the study (online supplemental figure 2). In a post hoc analysis of data from RA-BEAM, a decrease in the MPV was observed concurrently with the increase in overall mean platelet count (online supplemental figure 3).
Laboratory abnormality AEs related to platelet counts were reported in 51/3492 (1.5%) patients. Platelet-count related laboratory abnormalities led to permanent discontinuation during the placebo controlled treatment period for one patient (0.2%) in the baricitinib 2 mg and one patient (0.1%) in the baricitinib 4 mg treatment group. Permanent discontinuation due to platelet-related laboratory abnormalities was reported for 8/3492 patients (0.2%) in the all Bari-RA set (table 5).
Venous thromboembolism
Platelet increases at 2 weeks were not associated with venous thromboembolic events (VTE).26 Through the 19-month safety update, the highest post-baseline platelet count was ≥400×109/L for similar proportions of patients with (15/42, 35.7%) and without (1278/3435, 37.2%) VTE. Highest post-baseline platelet counts were ≥600×109/L for 3/42 (7.1%) patients with and 123/3435 (3.6%) patients without VTE (online supplemental figure 4).
Haemoglobin and related laboratory parameters
Anaemia at screening or baseline
To explore the relationship between age or gender and disease activity level (represented by hsCRP) with anaemia before baricitinib treatment, data were assessed at screening (for screen failures) or baseline (for randomised patients) using pooled data from two of the phase 3 studies conducted in csDMARDs-experienced patients. The incidence of anaemia, defined either based on gaLLN (see the Methods section) or Grade 1 (<6.2 mmol/L [10.0 g/dL]) increased with hsCRP levels above the upper limit of normal (ULN), and was highest in patients with hsCRP that was more than 10x the ULN (online supplemental figure 5). When categorised by age, differences were found among patients with hsCRP more than twice the ULN, of whom those younger than 45 years had the highest incidence of haemoglobin below the gaLLN (online supplemental figure 5).
To assess the incidence of anaemia by background csDMARDs, baseline data for randomised patients from the same two phase 3 studies were assessed. At baseline, anaemia (haemoglobin <gaLLN or anaemia of more than Grade 1) was most commonly present among patients taking MTX, with or without additional csDMARDs (online supplemental table 2).
On-treatment analyses
When assessed through up to 128 weeks, small declines from baseline in mean haemoglobin levels were observed for both the placebo and baricitinib dose groups at week 2 (placebo −0.15 mmol/L (0.24 g/dL), both baricitinib doses −0.12 mmol/L (−0.19 g/dL)) and again at week 14 (placebo −0.14 mmol/L (−0.23 g/dL), baricitinib 2 mg −0.18 mmol/L (−0.29 g/dL) and Bari 4 mg −0.22 mmol/L (−0.35 g/dL)) (figure 3). In patients receiving baricitinib, mean haemoglobin levels returned to baseline or above by weeks 48–52.
Changes in haemoglobin concentration over time were also assessed by quartiles of baseline hsCRP for patients receiving baricitinib 4 mg (online supplemental figure 6). For the lowest three quartiles of baseline hsCRP, baseline haemoglobin concentrations were roughly similar, and transient decreases in mean haemoglobin concentration with a return to baseline levels were seen. For the highest baseline hsCRP quartile, which had a lower mean haemoglobin concentration at baseline, mean haemoglobin concentration decreased transiently and then increased to levels comparable to the mean baseline haemoglobin levels in the other three quartiles. Within the highest baseline hsCRP quartile, there was a significant correlation between the increases in haemoglobin over time and the reduction in hsCRP observed during treatment (change from baseline to week 52, Pearson r=−0.30, p<0.001; from baseline to week 104, Pearson r=−0.24, p=0.039).
During the 24-week placebo controlled treatment period, the incidence of treatment-emergent abnormally low levels of haemoglobin (see the Methods section) was comparable between patients receiving placebo (25.8%) and baricitinib 4 mg (29.3%) and was lower among patients receiving adalimumab (16.9%, in the RA-BEAM study). The incidence was also similar across baricitinib doses when compared in the combined studies that included both baricitinib 2 mg (30.9%) and 4 mg (35.0%) treatment arms. Similarly, there was a low overall incidence of treatment-emergent shifts in haemoglobin from Grade 1 or 2 (haemoglobin ≥4.9 mmol/L [7.9 g/dL]) to Grade 3 or higher (haemoglobin <4.9 mmol/L) of 0.2% with placebo, 0.6% with baricitinib 2 mg and ≤0.2% with baricitinib 4 mg.
AEs related to decreased haemoglobin concentrations were reported in 174/3492 patients (5.0%). Study drug was discontinued permanently because of decreased haemoglobin/anaemia in two patients (0.4%) in the baricitinib 2 mg group and one patient (0.1%) in the baricitinib 4 mg group during the 24 week placebo-controlled period. Permanent discontinuation due to decreased haemoglobin/anaemia was reported for 16/3492 (0.5%) of patients in the all Bari-RA data set (table 5).
Erythropoietin- and iron-related measures in the JADN study
Erythropoietin concentrations, total iron, total iron-binding capacity (TIBC), red blood cell count and reticulocyte counts accompanying haemoglobin changes were analysed in the JADN study.27 Dose-dependent increases in erythropoietin levels were observed over 12 weeks in both the JADN 2 mg and JADN 4 mg baricitinib treatment groups (online supplemental figure 7). Mean red blood cell counts in JADN decreased slightly and remained stable in patients treated with either baricitinib 2 mg or 4 mg, similar to changes observed in subjects who received placebo (−1.0% for placebo at 12 weeks, −1.5% for baricitinib 2 mg and −5.0% for baricitinib 4 mg). For patients in JADN who were treated with either baricitinib dose, an initial decrease in reticulocyte count occurred at week 2. Among patients receiving baricitinib 4 mg, this initial decrease was followed by an increase in reticulocyte count that continued through week 12. Among patients treated with baricitinib 2 mg, reticulocyte counts returned to baseline at week 4, decreased again at week 8 and then returned to near baseline at week 12.
Statistically significant increases in total iron and TIBC were observed at week 12 in the JADN study with baricitinib 4 mg treatment, as compared to placebo (online supplemental figure 8).
DISCUSSION
Changes in haematological parameters, assessed across the baricitinib development programme, were generally small-to-moderate in magnitude and were often transient. The initial decrease in neutrophils is similar to those reported in studies of other JAK inhibitors for RA including tofacitinib, upadacitinib and filgotinib.11 28–31 While changes in other haematological parameters were generally small-to-moderate with all JAK inhibitors, there are differences in the patterns observed. Baricitinib treatment resulted in initial small transient increases in platelets, which then decreased towards baseline and remained within normal range, whereas treatment with tofacitinib or filgotinib resulted in moderate decreases in platelet counts within normal range, and upadacitinib had little impact on platelet counts.31–33 Baricitinib treatment resulted in initial reductions in haemoglobin within normal range which subsequently increased above baseline, while upadacitinib treatment had little impact on haemoglobin.33 Filgotinib treatment resulted in dose-dependent increases in haemoglobin, which plateaued after 12 weeks,31 and tofactinib treatment resulted in increases in haemoglobin, which plateaued after 12 months but were not dose-dependent.11
Serious infections, including tuberculosis, bacterial, invasive fungal, viral and other opportunistic infections, have been observed with JAK inhibitor use.34 While an increase in overall infections was observed among patients treated with baricitinib 4 mg, rates of serious infections (those resulting in hospitalisation, death or use of intravenous antibiotics) were similar to those in the placebo-treated groups.35 In the present study, lymphopenia was associated with a slightly higher rate of overall infection among patients treated with baricitinib 4 mg compared with that among patients receiving placebo, but serious infections were uncommon in patients with lymphopenia in both the placebo and in the baricitinib 2 mg and 4 mg treatment groups. A decrease in levels of neutrophils was observed with baricitinib dosing; however, serious infections were uncommon in patients with neutropenia, occurring in only one patient each (both with Grade 1 neutropenia) in the baricitinib 2 mg and 4 mg treatment arms. In RA-BEAM, a similar pattern of change in neutrophil count was observed in patients treated with baricitinib and adalimumab through 52 weeks.18
In the analysis of platelet counts over time, baricitinib (2 mg and 4 mg) was associated with transient increases in the mean platelet count at week 2, which subsequently remained slightly above baseline. In the exploratory analysis of patients treated with baricitinib 4 mg in RA-BEAM, a low MPV was observed,36 consistent with the hypothesis that reduced platelet clearance is the primary mechanism by which the platelet count increases. Reduced rates of platelet clearance would result in persistence of older, smaller platelets in the circulation and thus a lower MPV. Because older, smaller platelets may contain fewer granules, express fewer adhesion molecules on their surface, and activate more slowly,37 38 they would be expected to cause fewer VTE than larger platelets.39 Although in the overall dataset a highest post-baseline platelet count ≥600×109/L was observed more frequently in patients who had VTEs versus those who did not (3/42 (7.1%) vs 123/3435 (3.6%, respectively)), interpretation of this finding is limited by its low overall incidence.
Baricitinib was associated with early, small decreases from baseline in mean haemoglobin concentrations during the placebo-controlled periods, which returned towards baseline levels with extended treatment. The observed decreases in haemoglobin levels were seldom considered to be clinically relevant, since permanent drug discontinuation due to anaemia occurred infrequently. The initial reduction of haemoglobin, followed by subsequent increase towards baseline likely resulted from JAK2 inhibition, by decreasing erythropoietin signal transduction, while also reducing concentrations of proinflammatory cytokines, such as interleukin (IL)-6, and signal transduction through the IL-6 receptor, resulting in a diminished inhibitory effect on erythropoiesis.40
Patients in the highest baseline hsCRP quartile, with the highest burden of inflammation, had lower baseline haemoglobin concentrations that increased with baricitinib treatment. This increase in haemoglobin during baricitinib treatment correlated significantly with the decrease in hsCRP in those patients whose baseline hsCRP was in the highest quartile. IL-6 induces production of acute phase reactants, such as CRP and hepcidin.41 42 Hepcidin regulates iron homoeostasis, inducing inhibition of intestinal iron reabsorption and ferroportin-dependent iron mobilisation from macrophages.42 Increases in haemoglobin in patients with RA treated with the anti-IL-6 receptor antibody tocilizumab are associated with the reversal of iron sequestration by hepcidin.43 By inhibiting IL-6 receptor signalling,44 baricitinib may also facilitate iron mobilisation. Our observation of increased iron in the JADN study supports the hypothesis that reduction in proinflammatory cytokine receptor signalling through JAK1 inhibition is a possible mechanism for the improvements in haemoglobin concentrations observed with baricitinib treatment. These mechanisms may account for the observation that neither frequency nor severity of anaemia increased with long-term baricitinib use.
This descriptive study using post hoc analyses has limitations. Across the studies, entry criteria limited enrolment of patients with low disease activity. In addition, patients with haemoglobin <10 g/dL were excluded from participation, as were those identified by site investigators as having other significant comorbidities. The absence of such subjects from these clinical trials may limit the generalisability of the results. The lack of data on comorbidities, and the exclusion from randomised clinical trials of patients with certain comorbidities that otherwise are commonly seen in patients with RA in clinical practice and which might impact haematological parameters are limitations of this study. Finally, analyses of erythropoietin, reticulocytes, total iron and TIBC were derived from a single study in Japanese patients (JADN); findings might differ in other populations.
In summary, baricitinib treatment was associated with moderate decreases in neutrophils, and with transient increases in lymphocytes and platelets, which usually returned to baseline levels and remained stable up to 128 weeks. Changes in haemoglobin were generally small-to-moderate and mostly reversible. Changes in haematological parameters seldom resulted in permanent discontinuation of therapy.
Acknowledgments
Kent Steinriede of Syneos Health Morrisville, NC, USA, provided editorial support in preparation of this manuscript, funded by Eli Lilly and Company. Portions of this work were previously presented at the EULAR meeting, Madrid 14–17 June 2017 (Ann Rheum Dis 2017; 76(suppl 2) 513–14); the 62nd Annual General Assembly and Scientific Meeting of the Japan College of Rheumatology, Tokyo, 26–28 April 2018 (Mod Rheumatol 2018;28(suppl 2018) S292); and the American College of Rheumatology/ARHP 2018 Annual Scientific Meeting (Arthritis Rheumatol. 2018; 70(suppl 10). https://acrabstracts.org/abstract/mean-platelet-volume-changes-with-baricitinib-indicate-reduced-new-platelet-production-in-baricitinib-treated-rheumatoid-arthritis-patients).
REFERENCES
Footnotes
Twitter Jonathan Kay @RheumKay.
Contributors All authors met the following criteria for authorship: substantial contributions to the acquisition, analysis and/or interpretation of data for the work; contribution to drafting the work and/or revising it critically; giving final approval of the version submitted; and agreeing to be accountable for all aspects of the work.
Funding The studies were designed by the sponsors, Eli Lilly and Company and Incyte Corporation, with input from an advisory board that included authors of this article who were not employees of Eli Lilly and Company or Incyte Corporation.
Competing interests JK reports grants paid to the University of Massachusetts Medical School from AbbVie, Genentech, Gilead Sciences, Pfizer and UCB; and personal fees from AbbVie, Amgen, Alvotech Suisse AG, Arena Pharmaceuticals, Boehringer Ingelheim GmbH, Celltrion Healthcare Co., Janssen Biotech, Merck Sharp & Dohme Corp., Mylan, Novartis AG, Pfizer, Samsung Bioepis, Sandoz and UCB. MH reports grants and personal fees from Bristol-Myers Squibb K.K. and AbbVie Japan; grants from Eisai, Ayumi Pharmaceutical Co., Nippon Kayaku Co., Mitsubishi Tanabe Pharma Co., and Teijin Pharma; and personal fees from Eli Lilly and Company, Boehringer-ingelheim, Kissei Pharmaceutical Co., and Chugai Pharmaceutical Co.. JR, CD, MI, IdlT, YI, AC and CS were employees and shareholders of Eli Lilly and Company. TM was an employee of Syneos Health under contract to Eli Lilly and Company. EK reports grants and personal fees from AbbVie, Amgen, Gilead, Merck, Eli Lilly and Company, Pfizer; grants from PuraPharm; and personal fees from AstraZeneca Pharma, Bristol-Myers Squibb, Celltrion, Jannsen, Myriad Autoimmune, F. Hoffmann-La Roche & Co, Genentech, Sandoz, Sanofi Genzyme, Samsung Bioepsis, and UCB. RFvV reports research support and Grants from Bristol-Myers Squibb, GSK, Eli Lilly and Co., Pfizer, UCB Pharma. Consultancy, honoraria: AbbVie, AstraZeneca, Biotest, Celgene, GlaxoSmithKline, Janssen, Eli Lilly and Co., Novartis, Pfizer, Servier, UCB. JTG reports personal fees from AbbVie, Bristol-Myers Squibb, Eli Lilly and Co., and UCB, and grants from Pfizer. TWJH from the Department of Rheumatology LUMC has received research support/lecture fees/consultancy fees from Abblynx, Merck, UCB, Bristol-Myers Squibb, Biotest AG, Janssen, Pfzer, Novartis, Roche, Sanofi-Aventis, Abbott, Crescendo Bioscience, Galapagos, Nycomed, Boeringher, Takeda, Zydus, Epirus and Eli Lilly and Co. JMK is a consultant and shareholder of Corrona, LLC, and a consultant for AbbVie, Amgen, Bristol-Myers Squibb, Genentech, Gilead, GlaxoSmithKline, Eli Lilly and Co., Pfizer, Regeneron and Sanofi.
Patient consent for publication Patients were not consulted regarding the design of the studies.
Ethics approval All trials were conducted in accordance with the ethical principles of Declaration of Helsinki and Good Clinical Practice guidelines. The institutional review board at each investigational centre approved the study protocols. All patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the USA and European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data availabilty agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data availabilty environment. For details on submitting a request, see the instructions provided at www.vivli.org.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.