Article Text

Abstract
Objectives Besides adaptive immunity genes, genetic risk factors for psoriatic arthritis (PsA) include innate immunity loci, which suggests an autoinflammatory disease mechanism, at least in a subset of patients. Here, we aimed at investigating the autoinflammatory genetic background of PsA.
Methods A total of 120 patients with PsA visiting the outpatient clinics of the Hannover University hospital underwent targeted next-generation sequencing, searching for variations in genes linked with inborn errors of immunity classified as autoinflammatory disorders (AIDs). Deleteriousness of rare variants was evaluated through in silico analysis.
Results We found 45 rare predicted deleterious variants in 37 out of 120 (30.8%) patients with PsA. Relatively common were variants in AP1S3, PLCG2, NOD2 and NLRP12. All 45 variants were monoallelic and 25 of them, identified in 20 out of 120 (16.7%) patients, were localised in genes associated with autosomal dominant (AD) disorders. Detection of those variants is associated with pustular psoriasis or a coexisting inflammatory bowel disease (IBD).
Conclusions Approximately 30% of patients with PsA harboured at least one variant in a gene associated with an AID, suggesting an autoinflammatory disease mechanism. Detection of variants in genes linked to AD-AIDs may explain extra-articular manifestations of PsA, such as pustular psoriasis and IBD.
- immune system diseases
- inflammation
- polymorphism, genetic
- epidemiology
- arthritis, psoriatic
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Genetic susceptibility to psoriatic arthritis (PsA) is partially explained through innate immune loci, suggesting a link between PsA and autoinflammation.
WHAT THIS STUDY ADDS
This is the first study evaluating the genetic link between PsA and autoinflammatory disorders (AIDs).
The identification of genetic variants that associate with AIDs in patients with PsA provides evidence on the autoinflammatory pathomechanism of PsA.
In the subgroup of patients with variants in genes linked to AIDs, PsA could be a manifestation of a monogenic AID.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study alerts treating physicians for diagnosis of an underlying AID in patients with PsA, especially in case of a coexisting inflammatory bowel disease or pustular psoriasis.
The autoinflammatory aetiology of PsA may lead to re-evaluation of treatment of both cutaneous and articular inflammation, suggesting innate immunity targets, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interleukin-36 as likely therapeutic targets.
Introduction
Psoriatic arthritis (PsA) is a complex inflammatory disease of the joints associated with psoriasis.1 Beyond skin and varying articular features, inflammation in PsA can affect the entheses, the nails, the eyes and the gut.2 The fact that a family history of psoriasis represents a risk factor for the development of PsA, which has been integrated in the 2006 Classification for Psoriatic Arthritis (CASPAR) criteria,3 suggests the importance of genetic factors in the pathogenesis of PsA and its clinical spectrum.1 Genetic risk factors for PsA partially overlap with the ones of psoriasis.4 However, the heritability of PsA appears considerably higher than that of psoriasis, exceeding 80%, according to population-based studies that revealed a 30-fold to 49-fold relative risk in a case of a sibling with PsA.1 5 6 The latter suggests that within the clinical spectrum of psoriatic disease, genetic factors have a stronger influence on the development of arthritis as compared with a skin-only disease.7
Associations with human leucocyte antigen (HLA) class I alleles highlight the role of adaptive immunity, suggesting an autoimmune pathogenesis for PsA.8–11 The HLA-C*06 has been identified as the main genetic determinant conferring susceptibility to psoriasis.11 Despite its coassociation with PsA, the association with psoriasis is more robust and HLA-C*06 associates with a longer interval between the onset of skin disease and subsequent development of arthritis, which may suggest a rather protective effect on articular involvement.8–10 In contrast, within the HLA-B locus, HLA-B*27 and HLA-B*39 associate with a higher risk for developing PsA as well as with a shorter interval between the onset of psoriasis and the development of arthritis.1 8–11 Consistent with an autoimmune inflammation in PsA is the recently reported clonal expansion of CD8+ T cells in the synovial fluid of PsA.11 Further, diverse autoantibodies have been reported in PsA, although their pathogenicity has not been established.12 13 In addition to HLA, non-HLA innate immunity genes have been associated with psoriasis and PsA, suggesting the pathogenic relevance of innate immune dysfunction.1 Variants in genes involved in NF-κB activity, such as in TNFAIP3, encoding the protein A20, NFKBIA, encoding IκBα, which both negatively regulates NF-κB, as well as variants in CARD14, encoding the caspase recruitment domain-containing protein 14 (CARD14), which is involved in the initiation of the NF-κB signalling pathway, have been identified through genome-wide association studies (GWAS) to confer susceptibility for both psoriasis and psoriasis arthritis.14–16
Autoinflammatory diseases are an expanding group of heterogeneous conditions, characterised by inflammation, originating from a dysfunction of the innate immune system.17 The term ‘autoinflammatory’ was initially introduced to describe systemic inflammatory disorders that differ from autoimmune diseases in that high-titre autoantibodies and autoreactive T cells were absent.18 However, according to the current perception, detection of autoantibodies or T cell activation can be part of an autoinflammatory disorder, as long as the activation of the adaptive immune system is not driven from misrecognition of a self-antigen.19 So far, 56 monogenic inborn errors of immunity (IEI) resulting in autoinflammatory disorders are known.20 Their clinical spectrum comprises recurrent fever, mucocutaneous, neurological, gastrointestinal manifestations and rheumatic manifestations. Autoinflammatory pathomechanisms are increasingly recognised in common disorders, including atherosclerosis, gout and Alzheimer’s disease,21–23 and autoinflammation has been suggested to be involved in the pathogenesis of systemic rheumatic diseases, including spondyloarthritis and systemic lupus erythematosus (SLE).24 25
Previous genetic studies have suggested the autoinflammatory aetiology of PsA.14 15 Further, phenotypic traits of monogenic autoinflammatory disorders include features of the psoriatic spectrum, such as psoriasis, arthritis, uveitis and inflammatory bowel disease (IBD).19 Hence, we employed targeted next-generation sequencing (NGS), searching for variants in genes associated with monogenic autoinflammatory disorders in a cohort of patients with PsA, including subgroups of patients with variable articular and extra-articular manifestations.
Patients and methods
Study cohort
All patients visiting our rheumatology outpatient clinic between October 2020 and March 2021 were screened for the diagnosis of PsA. Diagnosis of PsA has been made according to the 2006 classification criteria for PsA of the CASPAR study group.3 Out of 3646 screened patients, 130 were diagnosed with PsA. A total of 120 of them with available consent and gDNA probe were enrolled in the study. Demographics, clinical and laboratory data were obtained from patients’ medical files. Those included all articular and extra-articular manifestations of the psoriatic spectrum,2 the age at onset of first psoriatic manifestation and patients actual and previous treatment.
Targeted NGS
Targeted NGS has been performed as described previously.26 Briefly, we generated a customised panel of genes associated with IEI grouped as diseases of immune dysregulation or autoinflammatory disorders (online supplemental table 1) with the help of Agilent’s web-based SureDesign application. Blood samples were collected in the immunology outpatient clinics of the Department of Rheumatology and Immunology of Hannover University Hospital. Genomic DNA was extracted by QIAamp DNA Blood Midi Kit (Qiagen) and quantified by Qubit dsDNA BR Assay Kit (ThermoFisher). DNA target enrichment was performed using SureSelect XTHS2 Target Enrichment System for Illumina Sequencing following the manufacturer’s instructions (Agilent’s user manual). Sequencing was performed on an Illumina MiSeq system using an Illumina v2 reagent kit following the manufacturer’s protocol. Data analysis was performed with the help of Agilent’s SureCall software.
Supplemental material
Variant filtering
The FastQ files were aligned to the human reference genome (UCSC hg19, GRCh37) and analysed using Agilent Technologies—SureCall software. Variants were selected according to criteria at the variant level: allele frequency (AF), variant annotation and potential functional effect. Using databases of variants (eg, dbSNP, 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD)) and disease-causing variants (HGMD, OMIM), we selected all private or rare variants with an AF <1%. Furthermore, we kept nonsense variants, variants affecting splice site, frameshift, in-frame indels, start or stop codon changes, as well as missense variants that were predicted deleterious by having a Combined Annotation-Dependent Depletion (CADD) score >15 and a Mutation Significance Cut-off (MSC) score below the CADD score.27–29 Pathogenicity of variants that fulfilled all aforementioned criteria has been additionally evaluated with the Sorting Tolerant From Intolerant (SIFT) algorithm,30 the MutationTaster,31 Provean32 and the Human Splicing Finder.33
Statistical analysis
For statistical calculation, we used GraphPad Prism V.5.00 (GraphPad, La Jolla, California, USA). Descriptive statistics are reported as median and IQR in case of continuous variables and as counts and percentages for dichotomous variables. Categorical variables were compared by the χ2 test. Non-categorical variables were compared with the Mann-Whitney test. All comparisons were two-tailed and p<0.05 was considered significant.
Results
Characteristics of patients with PsA
The characteristics of all 120 enrolled in the study, including PsA-associated manifestations and treatment, are summarised in table 1. The majority of studied patients had active psoriasis (96/120, 80%) at the time of sample collection. Plaque psoriasis was the most common type of psoriasis, diagnosed in 83 out of 120 (69.2%), followed by pustular psoriasis (13/96, 13.5%). A total of 9 out of 120 (9.4%) had nail psoriasis, which in all cases accompanied skin psoriasis, and mostly plaque psoriasis (8/9). Nearly all patients without psoriasis (ie, 23/120, 19.2%) had a family history of psoriasis. Overall, 38 out of 120 (31.7) patients had a family history of psoriasis and the majority of them, that is, 34 out of 38 (89.5%) had at least an affected first-degree relative. To our knowledge, all enrolled patients with PsA were unrelated. In addition, we documented all non-psoriatic manifestations of immune dysregulation, and identified 11 out of 120 (9.2%) patients with hypothyroidism, most commonly as a consequence of Hashimoto’s thyroiditis (8/11), 8out of 120 (6.7%) patients with a history of atopic disease, including atopic dermatitis (6/8) and asthma (4/8), 3 out of 120 (2.5%) patients with giant cell arteritis, 3 out of 120 (2.5%) patients with a history of erythema nodosum, 2 patients with a history of Raynaud’s phenomenon, 2 patients with vitiligo, 2 with a history of Sicca symptoms and 2 with oral aphthosis.
Characteristics of the patients with PsA
Detection of variants in genes associated with autoinflammatory disorders in patients with PsA
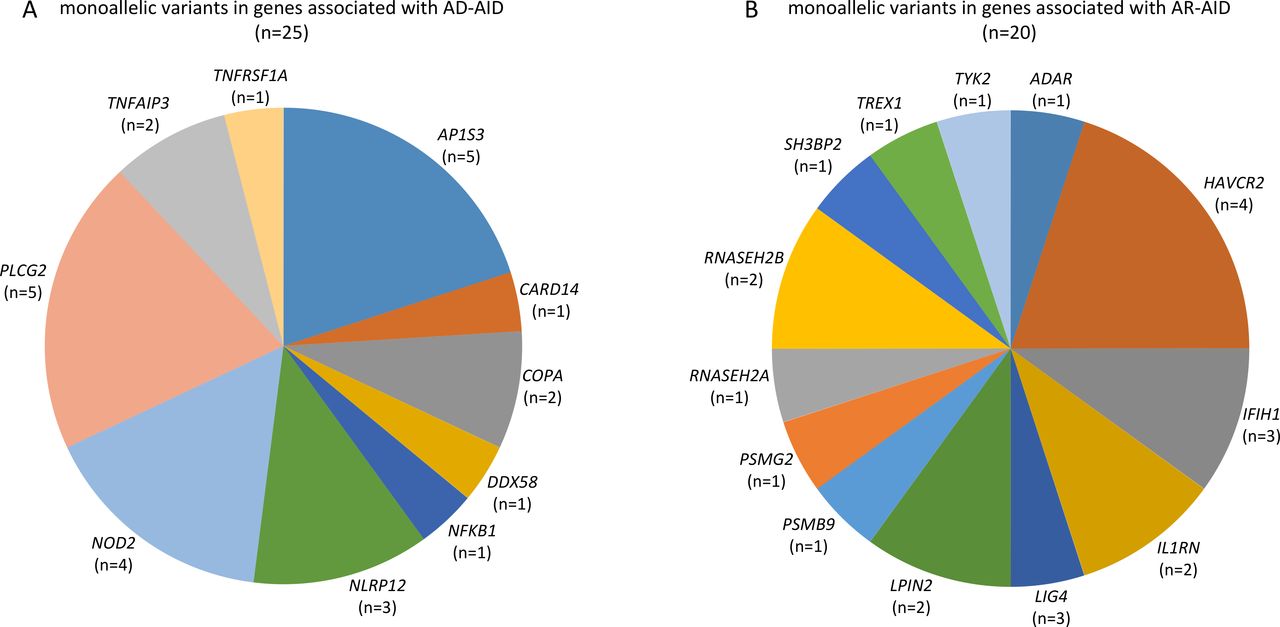
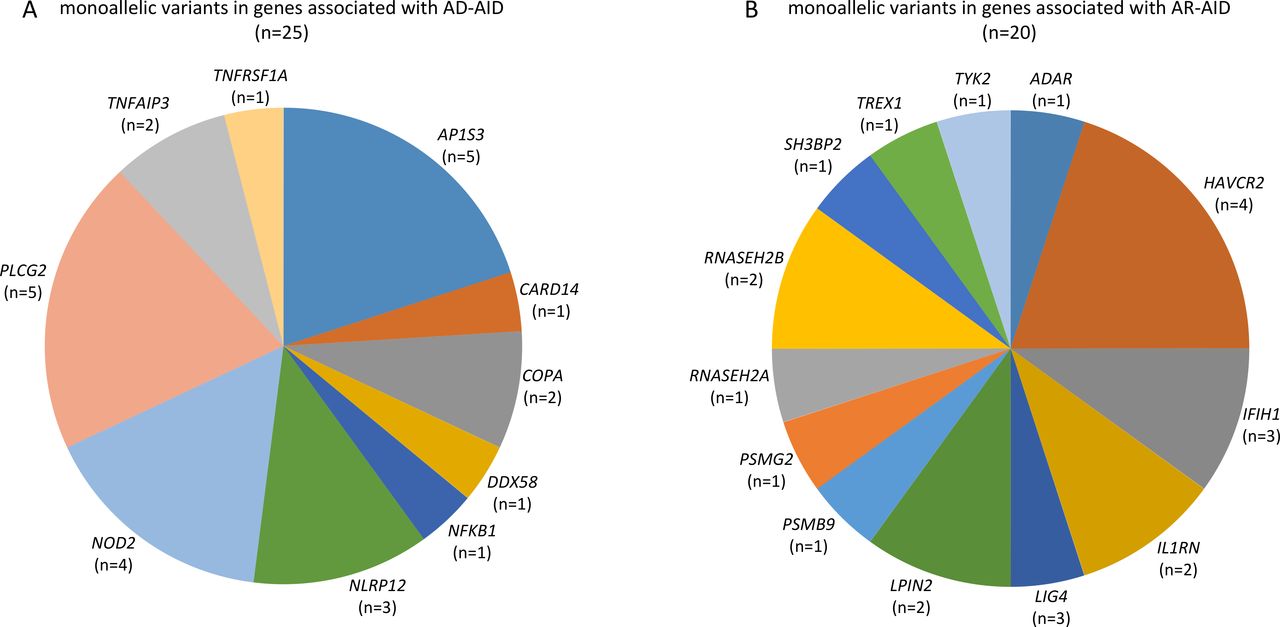
The employed targeted NGS approach included all genes, which were associated with IEI, classified as autoinflammatory disorders (group VII of the International Union of Immunological Societies (IUIS) classification of IEI) as well as genes associated with disorders from alternative groups, that have also been associated with autoinflammatory manifestations.20 Considering AF as well as measures of deleteriousness, such the CADD and the MSC score of each identified variant, we ended up with a total of 45 rare and likely deleterious germline variations (table 2), which were identified in 37 out of 120 (30.8%) patients (figure 1). All identified variants were monoallelic. A total of 25 variants in genes linked to an autosomal dominant (AD) disorder were detected in 20 out of 120 (16.7%) patients (figure 1 and figure 2A).

Summary of psoriatic manifestations (map, green boxes) and genetic findings (map, blue boxes for variant in genes associated with AD-AIDs and yellow boxes for variants in genes associated with AR-AID) in n=120 patients with PsA; each map column matches a studied patient (upper map summarises findings from patients 1 to 60 and the lower one from 61 to 120). AD-AID, autosomal dominant autoinflammatory disorder; AR-AID, autosomal recessive autoinflammatory disorder; IBD, inflammatory bowel disease; SpA, spondyloarthritis.
{kind=link}
{kind=link}
Summary of genes linked to autoinflammatory disorders, whose variations were detected in a cohort of patients with psoriatic arthritis: (A) genes linked to AD-AID and (B) genes linked to AR-AID. Variants in IFIH1 and TREX were listed under AR-AID, although mutations in either of these genes can also cause AD-AIDs. AD-AID, autosomal dominant autoinflammatory disorder; AR-AID, autosomal recessive autoinflammatory disorder.
Monoallelic variants in genes associated with monogenic autoinflammatory disorders or other inborn errors of immunity associated with immune dysregulation, identified in patients with PsA
Among those, 5 out of 120 (4.2%) patients were identified with a heterozygous variants in AP1S3 (figure 1). All those five patients harboured either the c.11T>G (p.Phe4Cys) or the c.97C>T (p.Arg33Trp) (table 2). Both the aforementioned AP1S3 variants have been previously reported as pathogenic and associated primarily with pustular psoriasis.34 35 Among patients with PsA harbouring an AP1S3 variant, four out of five had pustular psoriasis, whereas one was diagnosed with plaque psoriasis only (online supplemental table 2). In addition to psoriasis, those patients displayed variable arthritis, classified as a rather symmetrical polyarthritis with involvement of the finger joints in three patients and bilateral sacroiliitis in one of the five patients.
Further, we identified five rare variants in PLCG2 in four patients. PLCG2 encodes the phospholipase Cγ2 (PLCγ2), a signalling mediator primarily expressed by B cells, NK cells and mast cells.36 Monoallelic gain-of-function mutations in PLCG2 can cause a rare autoinflammatory syndrome, the autoinflammatory PLCγ2-associated antibody deficiency and immune dysregulation (APLAID), which causes early-onset dermatitis, ocular inflammation, enterocolitis, interstitial lung disease, arthritis and immunodeficiency.36 37 The four patients identified with PLCG2 variants displayed variable late-onset seronegative arthritis, which due to the coexistence of plaque psoriasis (3/4) or the family history of psoriasis in one of them has been classified as PsA (online supplemental table 3). Two of the patients also displayed mild hypogammaglobulinemia associated with a history of recurrent respiratory tract infections in one of them.
In addition, four NOD2 variants were identified in three patients. The NOD2 gene encodes an intracellular innate immune receptor and NOD2 mutations have been associated with chronic inflammatory disorders, that is, early-onset sarcoidosis, the Blau syndrome and Crohn’s disease.38 Two out of four patients harbouring a NOD2 variant displayed Crohn’s disease (online supplemental table 4). All other detected genetic variants are listed in table 2.
Features of PsA in patients with variants in genes linked to autosomal dominant AIDs
All psoriatic and non-psoriatic manifestations of immune dysregulation of the 20 out of 120 patients harbouring at least one variant in a gene associated with an AD-AID are presented in table 3. Among those pustular psoriasis was significantly more common in patients with a genetic variant associated with an AD-AID (5/20 vs 9/100, p=0.0419; table 4), which is mainly explained through the subgroup of patients with an AP1S3 variant. In particular, consistent with previous association of AP1S3 with pustular psoriasis,34 this psoriasis form was more commonly diagnosed among the subgroup of patients with an AP1S3 variant (4/5 vs 10/115, p<0.0001). Variants in AD-AID-associated genes were also more frequently detected among the subgroup of PsA patients with Crohn’s disease (3/20 vs 2/100, p=0.0079), which can be largely attributed to the NOD2 variants, which were detected in two out of five patients with Crohn’s disease.
Clinical data of PsA patients with monoallelic mutations in genes associated with AD-IEI/AID
Characteristics of psoriatic arthritis in patients with and without variants in genes associated with autoinflammatory disorders
Pharmacological therapies of patients harbouring variants in genes associated with AD-AID ranged from monotherapy with diverse non-steroidal anti-inflammatory drugs (NSAIDs) to conventional synthetic, targeted or biological disease-modifying antirheumatic drugs (DMARDs) and were similar to the ones employed to treat PsA in patients without such variants (online supplemental table 5). Patients with AD-AID-associated variants displayed higher serum levels of C reactive protein (CRP levels in patients with AD-AID-associated, indicating higher systemic inflammation that may be consistent with higher activity of PsA. However, rate of escalation/change of pharmacological therapy due to high disease activity was not significantly different between the aforementioned subgroups of patients.
Discussion
The phenotypic spectrum of PsA, its relatively high heritability and the fact that part of its genetic susceptibility is explained through innate immune loci suggested a link between PsA and AIDs. To investigate the latter, we searched for genetic variants in genes associated with monogenic AIDs in a cohort of patients with PsA and identified at least one rare predicted deleterious variant that could account for a monogenic AID in approximately 17% of sequenced patients, especially among patients with pustular psoriasis and IBD. These genetic findings suggest the autoinflammatory aetiology of PsA in this subgroup of patients.
Relatively common were AP1S3 variants, which are associated with pustular psoriasis. AP1S3 encodes a subunit of the adaptor protein complex 1 (AP-1), a heterotetramer mediating vesicular transport between the trans-Golgi network and the endosomes.39 Identified monoallelic variants in AP1S3 have been previously reported to have a loss-of-function effect and cause generalised or palmoplantar pustular psoriasis.34 35 Mechanistically, autoinflammation in case of AP1S3 loss-of-function has been attributed to abnormalities in autophagy, resulting in dysregulated NF-κB activation and consequently increased IL-1 and IL-36α signalling.35 To our knowledge, arthritis had not been previously associated with pathogenic AP1S3 variants. In the present study, the frequency of patients with both PsA and pustular psoriasis who harboured a deleterious AP1S3 variant is considerably higher than the previously reported frequency of such variants among patients with pustular psoriasis only (4/14, ie, 28.6% vs 7%–10%), whose evaluation was also based on German and European cohorts.40 41 The latter, together with the identification of a case with a deleterious AP1S3 variant, who had plaque psoriasis only, suggests the strong association of deleterious AP1S3 variants not only with pustular psoriasis, but also with arthritis. However, the role of deleterious AP1S3 variants in immune cells that could be relevant for the pathogenesis of arthritis, especially neutrophils that have been shown to display a relatively high AP1S3 expression,35 has not been demonstrated yet.
In addition, we have identified four patients with PLCG2 variants. Hypermorphic heterozygous variants in this gene, resulted in hyperactivation of PLCγ2, have been reported to cause multiorgan autoinflammation.37 Except for arthritis and dermatitis, there was no evidence of interstitial lung disease and ocular inflammation, which are common phenotypic traits of APLAID,36 37 in the four patients with PLCG2 variants. Autoinflammation due to gain-of-function variants in PLCG2 has been attributed to an enhanced activation of the NALP3 inflammasome.37 However, enhanced IL-1β production has been inconsistently reported in patients with APLAID and IL-1 inhibitors have been reported to fail in controlling ALPAID-associated autoinflammation.37 42 43 Plcg2 knock-in mice, which harbour gain-of-function missense mutations in Plcg2, display both arthritis and dermatitis,44 45 which resemble the phenotypic traits of APLAID. Despite the involvement of PLCγ2 in B cell activation, transfer experiments in aforementioned mice have demonstrated that cells of myeloid origin primarily account for their inflammatory phenotype, which is consistent with the notion of autoinflammation, although the exact mechanism of inflammation, including involved molecular pathways, have not been investigated yet.
In several cases, detection of variants in genes associated with autoinflammation could provide an explanation for the form of psoriasis and, in particular, for pustular psoriasis in patients with variants in CARD1446 or AP1S334 as well as for extra-articular manifestations, such as aphthous stomatitis in a patient with an TNFAIP3 variant, that may cause A20 haploinsufficiency47 or severe enterocolitis in a patient with a monoallelic variant in PLCG2, which associates with APLAID (table 3).37 In addition to aiding delineation of the clinical heterogeneity of PsA, detection of AID-associated variants and the identification of the autoinflammatory aetiology of disease in subgroups of patients might affect strategy to treat PsA and associated manifestations. Despite the modest benefit of treatment with anakinra in patients with PsA,48 in cases of disease due to genetic variants that enhance the activation of the NALP3 inflammasome and consequently the production of IL-1β and IL-18, blocking the latter cytokines may represent an attractive therapeutic approach. Further, given the enhanced spontaneous and induced production of IL-36 by keratinocytes from patients with loss-of-function mutations in AP1S3, inhibiting IL-36 may represent a therapeutic target, at least to treat pustular psoriasis in these patients.35 In case of several genes, including PLCG2, which was commonly affected in the present study, elucidation of the pathomechanism of autoinflammation is a prerequisite for the development of a targeted therapy.
Population-based studies revealed a high recurrence risk ratio for PsA among first-degree relatives, resulting in a heritability estimate of more than 80%.1 5 6 High heritability of PsA has been explained through genetic factors, which were primarily identified through large-scale GWAS. So far, genetic studies in the field of PsA have been based on the hypothesis-free approach of GWAS, which aims at detecting relatively common variants at a genome-wide level in large populations.4 49 50 Genetic alterations identified to confer disease risk through GWAS have suggested the pathogenic relevance of immunological processes, including antigen presentation, cytokines, innate immune pathways and T helper cell differentiation in PsA.4 49 With the advent and expanding availability of NGS technologies, the focus of genetic studies has shifted towards rare variants,51 which in the field of immunological diseases, led to the discovery of an increasing number of monogenic disorders, falling under IEI, and to the dissection of immunological syndromes, characterised by broad phenotypic variability, into monogenic disorders.20 Besides primary immunodeficiency disorders, in case of systemic rheumatic disorders, NGS led to the diagnosis of monogenic forms of SLE,52 seronegative vasculitis,47 53 chronic polyarthritis54 or spondyloarthritis, especially in the subgroup of patients with hypogammaglobulinemia.55
Our study has several limitations. As discussed above, despite using stringent selection criteria, including the rarity and pathogenicity prediction scores, we did not demonstrate the pathogenicity of identified variants, which may lead to an overestimation of the incidence of AID among patients with psoriatic arthritis. However, five patients were identified with previously reported pathogenic variants, all in AP1S3 and in several cases. As in case of several other groups of IEIs, incomplete penetrance and variable expressivity of reported pathogenic variants questions the strictly monogenic aetiology, suggesting the pathogenic effect of additional genetic modifiers, epigenetic and environmental factors,56 which have not been addressed in the present study. Lack of a control group of patients represents a major limitation of our study. However, the variant filtering strategy employed in the present study included selection of rare variants. Further, high rate of familial cases may have led to selection bias, overestimating the proportion of patients with predicted deleterious variants. Nonetheless, PsA displays high heritability5 6 and the rate of positive family history was comparable to the one reported in previous studies.57 58
In summary, the identification of genetic variants that are associated with AIDs in patients with PsA provides evidence on the autoinflammatory pathogenesis of PsA. At least in a subgroup of patients with variants in genes linked to AD-AID, PsA could be a manifestation of a monogenic AID.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the ethical committee of the Hannover Medical School (approval number: 5582; 8875_BO_K_2020). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank all nurses, physicians and documentation personnel of the outpatient clinics of the Department of Rheumatology and Immunology of the Hannover Medical School for collecting blood samples, informing the patients about the study and documenting patients’ medications.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors GS conceived and planned the study. GS took the lead in writing the manuscript. TW and TT significantly contributed to drafting and revision of the paper. GS and FA contributed substantially to data acquisition and interpretation, and performed the statistical analysis. GS, TW, MA and TT collected DNA samples and performed targeted NGS. All authors approved the final version. GS is responsible for the overall content as the guarantor.
Funding This project was primarily funded by the Rosemarie-Germscheid foundation. It was additionally funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2155 “RESIST” – Project ID 39087428 and the German network for multiorgan autoimmune diseases (GAIN, 01GM1910E).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.