Article Text

Abstract
Objective Shared epitope (SE)-coding DRB1 alleles are associated with bone erosion in several diseases, including rheumatoid arthritis (RA) and periodontal disease (PD), but the underlying mechanism is unknown. We have recently identified the SE as an osteoclast-activating ligand. To better understand the biological effects of the SE in vivo, here we sought to determine whether it can facilitate spontaneous bone damage in naïve mice.
Methods 3-month old naïve transgenic mice that carry the human SE-coding allele DRB1*04:01, or a SE-negative allele DRB1*04:02 were studied. Bone tissues were analysed by micro-CT, and the tooth-supporting tissues were studied by histology, immunohistochemistry and immunofluorescence. Serum biomarkers were determined by ELISA.
Results Transgenic mice expressing the SE-coding DRB1*04:01 allele, but not mice carrying the SE-negative allele DRB1*04:02, showed spontaneous PD associated with interleukin (IL)-17 overabundance and periostin disruption. Mandibular bone volumetric and mineralisation parameters were significantly lower in SE-positive mice, and alveolar bone resorption was significantly increased in these mice. SE-positive mice also had more slender tibiae, and their marrow, cortical and total areas were lower than those of SE-negative mice. Additionally, significantly increased serum IL-17, tumour necrosis factor-α and osteoprotegrin levels were found in SE-positive mice, while their receptor activator of nuclear factor κ-B ligand levels were significantly lower.
Conclusions A human SE-coding allele increases the propensity to spontaneous bone-destructive periodontal inflammation and skeletal bone damage in transgenic mice. These findings provide new insights into the previously documented but poorly understood association of the SE with accelerated bone erosion in RA and several other human diseases.
- Gene Polymorphism
- Rheumatoid Arthritis
- Bone Mineral Density
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key questions
What is already known about this subject?
The shared epitope (SE) is associated with several bone-damaging diseases, including rheumatoid arthritis (RA) and periodontal disease (PD).
PD is more common in RA.
Bone damage in RA affects both articular and extra-articular bone.
Although PD is known to associate epidemiologically with RA, and both conditions associate with the SE, the pathogenic role of the SE is unknown.
What does this study add?
Transgenic mice carrying a human SE-coding gene develop spontaneous, bone-destructive PD, implicating for the first time the SE as a direct pathogenic factor in PD.
SE-positive mice also had slenderer tibiae, and their marrow, cortical and total areas were lower than those of SE-negative mice.
Additionally, significantly increased serum interleukin-17, tumour necrosis factor-α and osteoprotegrin levels were found in SE-positive mice.
These findings could provide new mechanistic insights into the previously documented but poorly understood association of the SE with bone erosion several diseases.
The study could identify a new mechanism of RA–PD association.
How might this impact on clinical practice?
Once translated to human systems, these findings could help design preventive, early diagnostic and therapeutic intervention modalities for bone damage-associated diseases, including, but not limited to, RA and PD.
Introduction
Bone erosion is a devastating outcome in rheumatoid arthritis (RA) and periodontitis (PD), two common diseases that co-associate epidemiologically.1–3 Although the molecular mechanism of the association is unknown, it is worth noting that both conditions involve an interleukin (IL)-17-producing T helper (Th17) cell-dominated inflammatory process4 ,5 and osteoclast (OC)-mediated bone damage.6 ,7 Both RA8 and PD9 ,10 have been shown to associate with shared epitope (SE)-coding HLA-DRB1 alleles. Cigarette smoking, which synergises with the SE to increase disease risk in RA,10 has been shown to increase PD risk as well.11 ,12 Additionally, protein citrullination, which closely associates with RA in SE-positive individuals13 has been purported to play a role in PD.14
We have recently identified the SE as a ligand that binds to cell surface calreticulin and activates pro-osteoclastogenic signalling.15–28 Using cell-free synthetic ligands expressing the SE motif, we have demonstrated that the SE enhanced Th17 polarisation24 and directly activated OC differentiation.26 When administered in vivo to mice with collagen-induced arthritis, the SE ligand enhanced joint inflammation, increased the abundance of tissue and bone marrow OCs, and enhanced bone damage in arthritic joints.23 ,25 Thus, the SE is a signal transduction ligand that contributes directly to bone destruction in a disease model of RA induced by antigen immunisation.
To investigate whether the SE ligand affect bone biology under physiological conditions as well, here we have undertaken to determine whether naïve transgenic mice expressing a SE-coding HLA-DRB1 allele display bone abnormalities in two extra-articular areas of interest: jaws and tibiae. The findings indicate that SE-expressing mice display extensive bone-destructive PD along with skeletal bone abnormalities. These finding directly implicate the SE, for the first time, as a pathogenic factor in a spontaneous disease model, and suggest a potential new molecular mechanism for the well-documented but poorly understood higher incidence of erosive PD and diffuse skeletal bone damage in RA. The findings also provide a testable hypothesis concerning the reported association of the SE with bone damage in non-RA conditions.10 ,29 ,30
Materials and methods
Animals
Tg mice, expressing SE-positive HLA-DR4*04:01 or SE-negative HLA-DR4*04:01 human alleles31 ,32 were kindly provided by Dr Chella David, at the Mayo Clinic, and are referred to as DRB1*04:01 Tg and DRB1*04:02 Tg mice, respectively. The two mouse strains have a mixed (predominantly B6) genetic background, which is ∼99% identical between them. Experiments were carried out in 3-month-old male and female mice, weighing 19–25 g, housed in a specific pathogen-free, temperature-controlled room (25°C) with a 12-hour dark and 12-hour light cycle. A total of 20 naïve mice (no=5 in each group) were studied. Animals were allocated blindly to four experimental groups based solely on their gender and genotype. All protocols for mouse experiments were approved by the University of Michigan Unit for Laboratory Animal Medicine and by the University of Michigan Committee on Use and Care of Animals. Mice were maintained in accordance with all applicable federal, state, local, and institutional laws, regulations, policies, principles and standards (including accreditation) governing animal research.
Micro-CT
Maxillae and mandibles
Hemimaxilla and hemimandible samples were formalin-fixed and examined by a micro-CT system (µCT100 Scanco Medical, Bassersdorf, Switzerland) embedded in 1% agarose in a 34 mm diameter tube. The specimens were scanned with a voxel size of 18 µm, and scan settings of 70 kVp, 114 µA, 0.5 mm AL filter, and integration time 500 ms. To assess alveolar bone loss, the distances from the cemento enemal junction (CEJ) to the alveolar bone crest (ABC) were measured at four sites of second molars (mesiobuccal, distobuccal, mesiopalatal and distopalatal) in three-dimensional (3D) images viewed from buccal and palatal sides, with the assistance of the image analysis system (MicroView V.2.2 Advanced Bone Analysis Application, GE Healthcare Pre-Clinical Imaging, London, Ontario, Canada). Linear alveolar bone loss was determined by measuring the distances from the CEJ to the ABC were measured (interdental area between first and second molars). The measurements were repeated two times per site, and the results are presented as the distances in millimetres. To establish the residual amount of alveolar bone remaining in the interdental area between the first and second molar, the bone mineral density (BMD), bone volume fraction (BVF), bone mineral content (BMC), tissue mineral content (TMC) and tissue mineral density (TMD) were determined using a volumetric method, quantifying the structural bone based on microarchitectural parameters.
Tibiae
Specimens were scanned with a voxel size of 12 µm, 70 kVp, 114 µA, 0.5 mm AL filter and integration time 500 ms. The tibiae were reoriented with the mid-diaphysis parallel to the z-axis, and bone length was measured as the distance between the most proximal and distal transverse planes. Regions of interest (ROI) were located for cortical and trabecular parameters. A diaphyseal cortical ROI spanning 10% of total Tibia length was located midway between the distal growth plate and third trochanter. Parameters including cortical thickness, cross-sectional area, bending moment of inertia in the anterior–posterior direction, endosteal perimeter and periosteal perimeter were quantified with a commercially available software (MicroView V.2.2 Advanced Bone Analysis Application, GE Healthcare Pre-Clinical Imaging, London, Ontario, Canada).
A trabecular ROI covering 10% of total tibia length was located under the proximal growth plate. The inner cortical surface was defined with a splining algorithm. Trabecular metaphyseal bone was isolated with a more conservative algorithm for each specimen based on the bimodal distribution between marrow and bone.33 Parameters including marrow area, cortical area and total area were quantified using standard stereology algorithms (MicroView V.2.2). A 3D sphere fitting algorithm was also used to confirm the stereology data for trabecular bone.34
Immunohistochemistry
Tissues were fixed in 10% buffered formalin, decalcified, cut at 5 μm thickness, mounted on slides and air-dried at room temperature. Sections were deparaffinised, rehydrated and incubated with 3% hydrogen peroxide in methanol for 15 min at room temperature to eliminate endogenous peroxidase activity. Antigen retrieval was carried out at 95°C for 30 min by placing the slides in 0.01 M sodium citrate buffer (pH 6.0). The slides were then incubated with a primary rabbit polyclonal antibody for either IL-17A (Abcam #ab79056), periostin (Abcam #ab14041) or an isotype control (Abcam ab#27478) at 4°C overnight followed by secondary goat antirabbit (Abcam #ab6721). For immune detection, the avidin–biotin complex method was performed according to the manufacturer's instructions (TL-060-QHL—UltraVision Quanto Detection System HRP from Thermo Scientific). Colour development was achieved with 3, 3-diaminobenzidine. Photographs were taken using an Olympus BX60 upright microscope.
Immunofluorescence
Slides were stained with a primary rabbit polyclonal antibody for IL-17 as above, followed by secondary donkey antirabbit IgG antibody conjugated to Alexa Fluor 594 (Abcam #ab150076) and stored at 4°C until use. DAPI (4′, 6-diamidino-2-phenylindole, Sigma, #D9542) was used for nuclei staining. Quantification was carried out by a Biotek Cytation 5 instrument, using the Gen5Image+ software (Biotek).
Quantification of serological biomarkers
Serum concentrations of osteoprotegrin (OPG), IL-6, receptor activator of nuclear factor κ-B ligand (RANKL), tumour necrosis factor (TNF)-α and IL-17 levels were analysed using Quantibody Mice Custom Array (RayBiotech, Norcross, Georgia, USA), according to the manufacturer's specifications. Each sample was assayed in quadruplicate. An Innoscan 710 with MAPIX software (Innopsys, Chicago, Illinois, USA) was used to determine fluorescence intensities.
Statistical analysis
Data analysis was performed with unpaired t-test, using a GraphPad Prism software V.6. All data are shown as mean±SEM. Statistical significance was defined as p<0.05, and is denoted graphically as follows: *p<0.05; **p<0.005; ***p<0.0005.
Results
Spontaneous PD in SE-expressing mice
Given the reported association between RA and PD1–3 and between these two diseases and the SE,9 ,10 ,35 we sought to determine whether the SE associates with spontaneous PD. To this end we examined 3 months old naïve Tg mice expressing the SE-coding DRB1*04:01 allele, and compared them to control, naïve Tg mice expressing a non-SE-coding allele, DRB1*04:02.
Micro-CT-based 3D volumetric measurements (figure 1A) revealed a statistically significant bone loss in mandibles of SE-positive DRB1*04:01 Tg male mice, compared with the SE-negative DRB1*04:02 Tg control group. As shown, bone volume (p=0.008), BVF (p=0.003), BMD (p=0.0004), BMC (p=0.002), TMC (p=0.005) and TMD (p=0.004) were all significantly lower in SE-positive males, compared with SE-negative control male mice (figure 1A). In females, there were significant differences in TMD (p=0.0001) and BMC (p=0.02) between the SE-positive DRB1*04:01 Tg and SE-negative DRB1*04:02 groups, but not in other parameters (see online supplementary table SI).

Periodontal micro-CT analyses. (A) 3D micro-CT-based quantification of mandibular bone parameters in SE-positive DRB1*04:01 (black bars) and SE-negative DRB1*04:02 (gray bars) Tg mice. Values represent mean and SEM (n=5 per group). (B) Sagittal, coronal and axial 2D (left 3 columns) and 3D (right column) micro-CT images of mandibles from representative SE-positive DRB1*04:01 (upper panel) and SE-negative DRB1*04:02 (lower panel) Tg male mice. 2D, two-dimensional; 3D, three-dimensional; ABC, alveolar bone crest; AP, anteroposterior; BV, bone volume; BMC, bone mineral content; BMD, bone mineral density; CEJ, cemento enemal junction; BVF, bone volume fraction; SE, shared epitope; TMC, tissue mineral content; TMD, tissue mineral density.
supplementary tables
In maxillae, there were significantly lower TMD readings in males (p=0.0009) and females (p=0.02) in the SE-positive DRB1*04:01 Tg as compared with SE-negative DRB1*04:02 Tg mouse groups, but not in other parameters (see online supplementary table SI). In 2D readings (see online supplementary table SII), male mandibles (figure 1B) and maxillae and female mandibles all showed higher linear distances between CEJ to the ABC in SE-positive DRB1*04:01 Tg, as compared with SE-negative DRB1*04:02 Tg mice; however, these differences did not reach statistical significance (see online supplementary table SII).
Histological examination of SE-positive Tg mice mandibles demonstrated increased periodontal inflammatory infiltrates, as compared with SE-negative Tg mice (figure 2A). Periostin is normally expressed in the periodontal ligament and other fibrous connective tissue36 and plays a key role in maintaining periodontal stability in relation to tooth mechanical function.37 Disruption of the periodontal ligament with irregular periostin is a common finding in PD. As can be seen in figure 2A, periodontal tissues of SE-positive DRB1*04:01 Tg mice displayed disrupted periodontal ligaments as indicated by an irregular distribution of periostin. In contrast, in the SE-negative DRB1*04:02 Tg mice periodontal tissues showed a well-organised ligament with normal distribution of periostin.
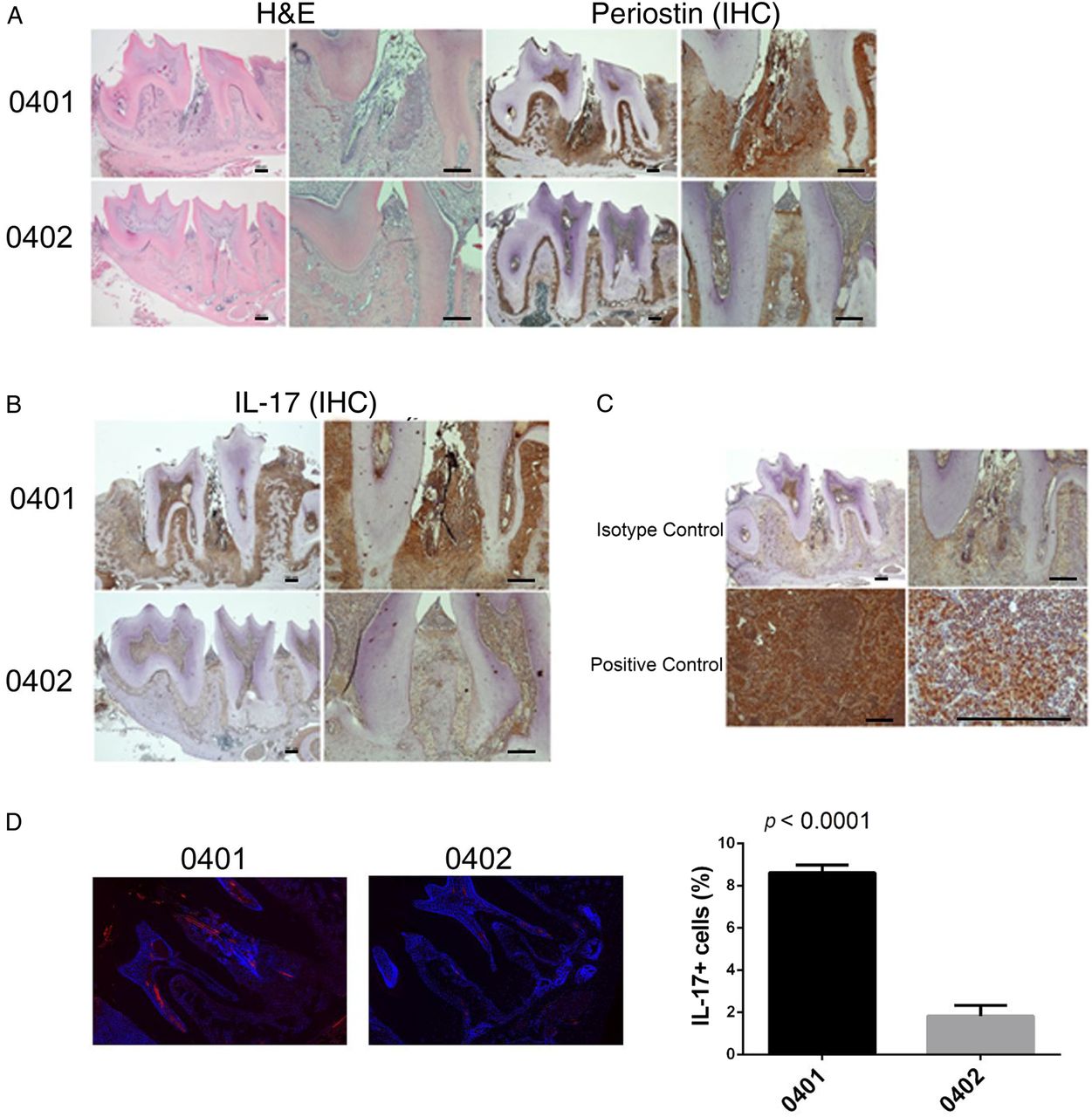
Spontaneous PD in DRB1*04:01 mice. (A) Representative mandibular tissue sections stained with H&E (left) show intense inflammatory infiltrate (left). IHC staining for periostin (right) show disruption of the periodontal ligament in SE-positive, DRB1*04:01 Tg mice (top panel). SE-negative DRB1*04:02 Tg mice (bottom panel) show normal histology. (B) IHC staining for IL-17A showing markedly increased abundance of the cytokine in mandibular periodontal tissue of SE-positive, DRB1*04:01 Tg mice (top panel), compared with SE-negative, DRB1*04:02 Tg mice (bottom panel). (C) Isotype-matched antibody control shows negative IHC staining (top panel). A positive control tissue (draining lymph node—10× and 40×) shows the expected abundance of IL-17. In H&E and IHC, images of low (4×, left) and high (10×, right) magnification are shown. Horizontal bars represent 100 µm. (D) Identification of IL-17-positive cells in periodontal tissues by immunofluorescence. Red fluorescence represents IL-17; blue fluorescence (DAPI) identifies nuclei. Boxed images show higher magnification images of regions identified by white arrows. Bar graph on the right depicts mean and SEM of IL-17-positive cells in SE-positive, DRB1*04:01 Tg mice (black) and SE-negative, DRB1*04:02 Tg mice (gray), as quantified by a Biotek Cytation5 instrument. DAPI, 4', 6-diamidino-2-phenylindole; IHC, immunohistochemistry; IL, interleukin; SE, shared epitope; PD, periodontal disease.
Immunohistochemistry staining for IL-17A showed higher abundance of the cytokine in SE-positive DRB1*04:01 mice compared with SE-negative DRB1*04:02 mice (figure 2B). Isotype-matched controls showed negative results, while a positive control tissue (inflammatory lymph node) showed strong staining (figure 2C). The overabundance of IL-17A in SE-positive mice was confirmed by immunofluorescence-based quantification (figure 2D). As can be seen, the SE-positive DRB1*04:01 Tg mice had a significantly higher percentage of IL-17A-expressing cells (8.613±0.7410%) as compared with SE-negative DRB1*04:02 Tg mice (1.826±1.024%; p<0.0001).
Long bone abnormalities in SE-positive mice
While SE-positive mice showed no spontaneous articular bone changes, they had profound long bone abnormalities. Representative tibial micro-CT images of SE-positive and SE-negative mice are shown in figure 3A. As table 1 specifies, total tibial area in SE-positive DRB1*04:01 males (0.65±0.03 mm2) was 20.6±0.086% lower than the area in SE-negative DRB1*04:02 male group (0.86±0.03 mm2; p=0.004). In SE-positive females, the total area (0.63±0.01 mm2) was 14.9±0.0026% lower than in the SE-negative group (0.74±0.02 mm2; p=0.005). SE-positive DRB1*04:01 male tibiae also showed significantly reduced marrow (p=0.002) and cortical (p=0.025) areas compared with SE-negative DRB1*04:02 males. In females, the SE-positive DRB1*04:01 group showed reduced tibial marrow (p=0.002) and total areas (p=0.005); however, the cortical area in females was not significantly different between SE-positive and SE-negative mice (table 1). Noteworthy, the differences between the SE-positive and SE-negative groups were not attributable to body mass differences, as weights did not differ significantly between SE-positive DRB1*04:01 and SE-negative DRB1*04:02 cohorts for either males or females (table 1). Different from the aforementioned differences, the trabecular bone was not affected by the mouse SE status (see online supplementary table SIII).
Tibiae–cortical bone parameters

Tibial micro-CT analysis. (A) Representative 2D (upper panel) and 3D (lower panel) tibiae micro-CT images of SE-positive DRB1*04:01 (left column) and SE-negative DRB1*04:02 (right column) mice. (B) Tibial mineral density data of SE-positive DRB1*04:01 (black bars) and SE-negative DRB1*04:02 (gray bars) mice. (C) Male (left) and female (right) robustness data of the SE-positive DRB1*04:01 (black dots) and SE-negative DRB1*04:02 (gray dots) mouse groups. Values in (B) and (C) represent mean and SEM (n=5 per group). 2D, two-dimensional; 3D, three-dimensional; SE, shared epitope.
There was also a difference found in TMD between SE-positive and SE-negative mice. As figure 3B demonstrates, DRB1*04:01 male tibiae showed significant lower TMD (p=0.0001) in comparison to the SE-negative group. The female SE-positive group also showed significant lower in TMD (p=0.0441) in comparison to the SE-negative group (figure 3B).
The SE status affected bone robustness as well. Robustness is defined as total cross-sectional area (Tt.Ar)/tibial length (Le) to be consistent with the fact that cross-sectional size increases proportional to the square of bone width (ie, area) during growth.38 The robustness (Tt.Ar/Le=total area) was found to be significantly lower in SE-positive males (p=0.0018) compared with the SE-negative control group, and a similar pattern was found in females (p=0.0045; figure 3C).
Serological markers
SE-positive and SE-negative mice differed in their serum cytokine levels (figure 4). As can be seen, TNF-α, IL-17 and OPG levels were all significantly higher in SE-positive DRB1*04:01 compared with SE-negative DRB1*04:02 mice (p=0.0148, 0.0253, and 0.0323, respectively). Interestingly, RANKL was significantly lower in SE-positive in comparison to SE-negative mice (p=0.03; figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serological markers. Serum concentrations of TNF-α, IL-17, RANKL and OPG in SE-positive DRB1*04:01 (black bars) and SE-negative DRB1*04:02 (gray bars) mice. Values represent mean and SEM (n=5 per group). IL, interleukin; OPG, osteoprotegrin; RANKL, receptor activator of nuclear factor κ-B ligand; SE, shared epitope; TNF, tumour necrosis factor.
Discussion
This study demonstrates that Tg mice carrying a human SE-coding HLA-DRB1*04:01 allele develop spontaneous erosive PD and skeletal cortical bone and marrow abnormalities, along with abnormal levels of inflammatory markers. The pathology is allele-specific, since no such aberrations were seen in control Tg mice carrying the HLA-DRB1*04:02 allele, which codes a DRβ chain that differs by only a few amino acid residues from the SE-positive chain. To the best of our knowledge, this is the first spontaneous disease model in SE-expressing Tg mice.
The SE, the most significant genetic risk factor for RA, was identified almost 30 years ago,8 but its functional role remains unclear. The prevailing hypothesis posits that the SE sequence, possibly together with one or two HLA-DR groove amino acid residues,39 ,40 may enable presentation of arthritogenic antigens to T cells. However, in addition to the fact that the identity of such putative antigen is unknown, the antigen presentation hypothesis as a sole aetiological basis for RA suffers from several additional important shortcomings, as discussed elsewhere.26 ,41
In previous studies, we have demonstrated that the SE acts as a signal transduction ligand that facilitates Th17 and OC differentiation. When administered to mice with experimental arthritis, synthetic SE ligands, which are incapable of any antigen presentation, facilitated disease severity and increased Th17 polarisation and OC-mediated bone damage.23–25 Directly relevant to the present study, naïve Tg mice carrying the SE-coding allele HLA-DRB1*04:01 showed higher propensity to osteoclastogenesis and bone degradation activity ex vivo.24 ,25 Thus, our previous studies indicate that independent of its putative role in antigen presentation, the SE may contribute directly to arthritis severity as a signal transduction ligand that facilitates an OC-mediated bone-destructive pathway.
The findings of this study lend support to a bone remodelling effect of the SE ligand in physiological conditions as well. As shown here, SE-expressing mice spontaneously developed severe Th17-dominated PD, associated with bone destruction. This happened without intentional exposure to any infectious, chemical or mechanical stimuli. The fact that the pathology found here was limited to extra-articular bone, while joint tissues were spared, suggests that a specific immune response to joint antigens is unlikely to be the underlying mechanism of the SE effect in these mice.
It is noteworthy that PD and RA have been previously found to co-associate.1–3 One of the hypotheses postulates that PD-associate pathogenic bacteria, such as Porphyromonas gingivalis may be the culprit, due to their ability to facilitate protein citrullination.42 However, conclusive evidence to substantiate this cause–effect relationship is yet to be found. The findings of this study suggest that irrespective of whether such putative cause–effect scenario plays a role, the SE facilitates widespread bone damage processes.
The bone effects of the SE extended beyond the periodontal site and included skeletal tissues. The reduced tibial total area in SE-positive mice suggests suppression of periosteal expansion during growth. This effect was seen in both sexes, albeit slightly more severe in males. The reduced cortical area in the males, but not females, suggests sex difference in mass accumulation. Additional studies examining mice at multiple time points are warranted to determine whether the reduced mass in males was a result of altered bone growth or excessive resorption (loss). The mechanism of SE-associated reduced periosteal expansion is unclear, but may be linked to our previous findings of OC activation and proinflammatory effects of the SE ligand. Together, the reduced external size, cortical mass and robustness would suggest that similar to patients with RA,43 ,44 SE-positive mice might have reduced bone strength.
It has been previously found that bone damage in RA involves articular, periarticular and systemic skeletal tissues.45 The reasons for this widespread bone damage are unclear. In this study, we demonstrated substantial skeletal bone damage in SE-expressing Tg mice. This finding may suggest that the pathogenesis of skeletal bone damage in this disease might be, at least partly, genetically determined by the SE ligand effect, rather than solely due to inflammation, as is commonly postulated.
Although SE-associated PD and bone pathology affected both male and female mice, there was a slightly greater predilection for such effects in males. The reasons for this apparent gender-biased effect are presently unclear. The trends were similar in both genders, but we cannot rule out that sex hormones may have a direct contributory role. Alternatively, the observed gender-associated differences could be related to secondary gender-associated factors. For example, gender-associated mechanical factors, such as body mass, mobility or mastication differences between males and female could have partially contributed to the observed differences. Further research is required to better determine whether and why PD and skeletal damage are disparately affecting male and female SE-expressing Tg mice.
Consistent with the previously documented proinflammatory effects of the SE ligand17 ,23–25 in this study we demonstrate that Tg mice expressing the SE-coding allele had higher constitutive serum levels of TNF-α and IL-17. Additionally, periodontal tissues in SE-positive mice displayed a significant overabundance of IL-17-positive cells in situ. Counterintuitively however, RANKL levels were lower, and OPG levels were higher in SE-positive mice. The reason for this finding is unknown; however, it may be consistent with our previous observations that SE ligand-activated OC differentiation is RANKL-independent, as the SE ligand could induce such differentiation even in the absence of RANKL,25 and our recent transcriptome analyses suggest that SE-activated and RANKL-activated osteoclastogenesis are mediated by substantially non-overlapping pathways (unpublished). Thus, it is tempting to speculate that the observed increased OPG levels in SE-expressing Tg mice might signify a defence response to other, RANKL-independent biochemical cues of excessive bone damage.
Finally, it should be noted that gut microbiota of SE-positive DRB1*04:01 Tg and SE-negative DRB1*04:02 Tg mice have been previously shown to differ.46 Whether or how such a purported difference could affect the findings of the present study is open to speculations. One future approach to answering this question would be to determine whether microbiota from susceptible, SE-positive HLA-DRB1*04:01 mice can transfer the bone phenotype to resistant, SE-negative HLA-DRB1*04:02 mice. Be that as it may, our previous studies have shown that the SE ligand directly activates OC-mediated bone damage in vitro in cell lines, independent of microbiota or other in vivo, strain-dependent or environmental influences.
In summary, we demonstrate here that Tg mice expressing a SE-coding HLA-DRB1 allele develop spontaneous erosive PD, skeletal abnormalities and serological evidence of a systemic inflammatory state. Molecular characterisation of the mechanisms involved could advance our understanding of the impact of the SE on bone remodelling in health and disease.
Acknowledgments
JH has been supported by the Eleanor and Larry Jackier Research Award from the University of Michigan—Israel Partnership for Research programme, and by grants from The National Institute of Dental and Craniofacial Research (R21 DE023845) and The National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR059085).
References
Footnotes
Contributors PG and SLV conducted experiments and acquired data. PG, HFR, KJJ and JH designed the research study, analysed data and wrote the manuscript.
Funding Eleanor and Larry Jackier Research Award from the UM—Israel Partnership for Research programme and by grants The National Institute of Dental and Craniofacial Research (R21 DE023845) and The National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR059085).
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests None declared.
Ethics approval All protocols for mouse experiments were approved by the University of Michigan Unit for Laboratory Animal Medicine and by the University of Michigan Committee on Use and Care of Animals.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.