Article Text

Abstract
This review summarises recent evidence about the interaction between bone, the immune system and cartilage in disabling conditions such as osteoarthritis, rheumatoid arthritis and spondyloarthritis. These topics have been recently discussed at the ‘OsteoRheumatology’ conference held in Genoa in October 2014. The meeting, at its 10th edition, has been conceived to bring together distinguished international experts in the fields of rheumatic and metabolic bone diseases with the aim of discussing emerging knowledge regarding the role of the bone tissue in rheumatic diseases. Moreover, this review focuses on new treatments based on underlying the pathophysiological processes in rheumatic diseases. Although, a number of issues still remain to be clarified, it seems quite clear that in clinical practice, as well as in basic and translational research, there is a need for more knowledge of the interactions between the cartilage, the immune system and the bone. In this context, ‘OsteoRheumatology’ represents a potential new discipline providing a greater insight into this interplay, in order to face the multifactorial and complex issues underlying common and disabling rheumatic diseases.
- Osteoarthritis
- Rheumatoid Arthritis
- Spondyloarthritis
- Treatment
- Osteoporosis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The basic idea underlying OsteoRheumatology is the importance of the crosstalk between inflammation, cartilage and subchondral bone in disabling conditions such as rheumatoid arthritis (RA) and osteoarthritis (OA). The growing interest on the topic led to significant progresses during the past 20 years, resulting from the continuous efforts of clinical and basic researchers worldwide.
Basic and clinical research focused on the interactions between the bone tissue, primarily the subchondral bone, the bone cells (osteoblasts, osteoclasts and osteocytes), the immune system and the cartilage in several rheumatological diseases, such as OA, spondyloarthritis (SpA) and RA. In this context, the bone damage as an early manifestation of arthritides, the systemic skeletal involvement in RA, the crucial role played by subchondral bone in the pathogenesis and progression of OA, the pathophysiology of glucocorticoid damage to the bone tissue and the potential beneficial effects of bisphosphonates and newly approved agents such as denosumab and biologics have been the matter of the study and debate in the among basic/clinical investigators in the past decade.
The growing awareness about the relevant interactions between the bone, the cartilage and the immune system in a number of rheumatic diseases encouraged the organisation of the first ‘Bone Involvement in Arthritis’ International Meeting (held in Venice in 2004), with the aims of bringing together distinguished international experts in the fields of rheumatic and metabolic bone diseases and discussing emerging evidence regarding the role of the bone tissue in rheumatic diseases. In 2011, the meeting changed its name to ‘OsteoRheumatology’, thus implying the necessity of giving a more clear definition to topics presented and issues raised.
This brief overview summarises the recent insights from the last two meetings, which took place in Genoa (Italy) during the years 2013 and 2014. The programme was organised by a scientific committee, chaired by Professor Gerolamo Bianchi.
Bone involvement in OA
OA is the most common joint disorder and is the major cause of disability in the adult population.1 Although cartilage destruction is the hallmark of OA, it is now well established that OA is not only a disorder of cartilage homeostasis but a whole-joint disease involving all of the articular tissues, including the subchondral bone and synovial membrane.2 More recently, some authors have extended this concept claiming that systemic factors such as low grade systemic inflammation (metabolic syndrome) or oestrogen deficiency are also involved in OA pathogenesis.3–5 For example, in women, oestrogen-deficiency has a strong influence on subchondral bone integrity and metabolism.4 In addition, there is evidence that, oestrogen-deficient animals develop slight but relevant histological changes in the healthy knee cartilage6 and osteoporosis aggravates OA progression and severity in the OA-induced model in rabbit.6 ,7
The role of subchondral bone in OA
The typical skeletal changes of OA are increased cortical plate thickness, flattening and deformation of the articular contour, bone marrow lesions, decreased subchondral trabecular bone mass, tidemark duplication and osteophyte formation.8 These changes affect the biomechanical properties of the overlying joint cartilage and intertwined biological relationship. The presence of subchondral bone stiffness may alter its viscoelastic proprieties and produce a loss of subchondral bone shock absorbing capacity with extra mechanical load.9–11 Several biological events confirming the presence of increased subchondral bone remodelling have been described in OA using different techniques, from scintigraphy to biomarkers. During progression of OA, subchondral turnover appears to be increased 20-fold compared to normal bone turnover.12 ,13 The increased subchondral bone remodelling in OA can give two different phenotypes: sclerosis or osteoporosis, and both conditions may coexist in the same patient. Uneven mechanical overload on the osteochondral junction can induce a sclerotic phenotype, meanwhile post-menopausal global osteoporosis or other factors such as ‘stress shielding’ or synovial inflammation can produce a subchondral bone osteoporosis phenotype. Despite increased bone volume fraction, subchondral bone in OA is hypomineralised and of inferior quality compare to normal bone tissue.14 ,15 It has been suggested that hypomineralisation of the collagen matrix of the OA subchondral bone is due to the presence of atypical osteoblasts with a peculiar phenotype which produce an abnormal homotrimeric type I collagen with low affinity for calcium.16 OA osteoblasts produce increased levels of alkaline phosphatase, osteocalcin, interleukin (IL)-6, IL-8, PG (prostaglandin)-E2, transforming growth factor (TGF)-β and insulin growth factor-1 (IGF-1). The local accumulation of IGF-1, which is a potent stimulator of bone formation, contributes to the subchondral bone sclerosis in OA.17 While IL-6, PGE2 and receptor activator of nuclear factor κ-B ligand (RANKL) may be responsible for increased number of osteoclasts in OA and bone resorption.
Recent studies have identified the central role of osteocytes in producing skeletal adaptive changes in OA in response to mechanical load. Osteocytes function as the bone mechanosensor and regulate bone remodelling by production of molecules that control both osteoclast and osteoblast differentiation and activity.18 The pathway that is involved in the osteoblast regulatory process by the osteocytes is the Wnt β-catenin pathway. In animal models increasing loading downregulates sclerostin production in osteocytes resulting in increased bone formation.19 ,20 Sclerostin expression is decreased in sclerotic subchondral bone in patients with OA and increased Wnt signaling in subchondral bone contributes to increased bone formation.21 ,22 In contrast, unloaded osteocytes are able to produce RANKL,23 ,24 which enhances bone resorption. The damage to the subchondral bone matrix, in addition to mechanical loading, modulates the activity of the osteocytes and this process likely accounts for the development of the bone marrow lesion.25 Chondrocytes can also contribute to subchondral bone remodelling by producing RANKL. RANKL has been shown to be expressed in chondrocytes obtained from patients with knee OA, especially in the deep layer,26 and in cartilage of instability-induced OA animal models.27 RANKL and inflammatory factors released by articular cartilage may contribute to subchondral bone deterioration through increased bone remodelling in OA. The interaction between subchondral bone and cartilage is under investigation. Interestingly, in an in vivo cyclical compression OA animal model, cartilage degradation corresponds to progressive thickening of subchondral cortical bone with development of osteoporotic changes in the trabecular bone demonstrating a cross-talk between these two tissues in OA.28 Sanchez et al17 have demonstrated in vitro that osteoblasts from sclerotic zones of subchondral OA through IL-6 production, downregulate, aggrecan synthesis and upregulate MMPs expression by chondrocytes. Microcracks identified at the bone/cartilage junction and blood vessels invasion allow the exchange of biochemical factors produce by osteoblasts between the bone and overlying cartilage.16 The angiogenesis at the osteochondral junction is associated with hypertrophic differentiation of chondrocytes, which synthesize high levels of vascular endothelial growth factor (VEGF) and bone sialoprotein, two factors promoting endothelial cell proliferation and migration.29 In addition to the cytokines that regulate bone formation, the interaction of osteoclasts with the bone substrate also provides a potent signal to induce the osteoclast differentiation.30 Mechanisms involving subchondral bone in OA are summarised in figure 1.

Different mechanisms may produce different phenotypes in osteoarthritis subchondral bone via increased subchondral bone remodelling. The figure summarises the role played by sclerostin, Wnt pathway, receptor activator of nuclear factor κ-B ligand (RANKL) and other inflammatory cytokines in the pathogenesis of the different osteoarthritis subchondral bone phenotypes.
New treatments in OA
The actual treatments recommended for OA include paracetamol, non-steroidal anti-inflammatory drugs (NSAIDs) and opioids. No disease-modifying drugs are available to date for this pathology.31 Treatment of OA is not only a question of targeting the right molecule but also it is important to identify the proper subset of patients to treat considering the different phases of the disease. Moreover, different joint tissues can be involved in OA pathogenesis, including the synovium, cartilage and subchondral bone and they can be targeted by different pharmaceutical agent. Presently, OA is regarded as a heterogeneous disease, with different phenotypes. As a consequence, the next step is to investigate new treatment options in clearly defined clinical subsets within the broad OA population.
Targeting pain
Pain is the hallmark symptom of OA and the major reason for total joint replacement. Pain after inactivity, stiffness and use-related pain are common, but rest and night pain particularly occur in severe OA.32 ,33 Different patterns of pain have been identified connected with different stages of OA. In the early phases of the disease, pain is related to activity and becomes more constant over time. In the late stages there is ‘background pain’ interspersed with unpredictable intense pain, which highly impacts the quality of life and the participation in social and recreational activities.34 Sometimes, pain is more severe than other times, probably related to inflammation.
Cartilage is not innervated, but other joint tissues can be a source of pain such as the meniscus, synovium, capsule, ligaments and periosteum. Moreover, in severe OA there is vascular and neural invasion at the bone–cartilage junction. An understanding of the pathophysiological mechanisms of pain in OA is essential to developing new therapeutic targets.35 Activation of nociceptive pathways is a physiological response to tissue injury and damage, and it is vital for the organism.36 In the presence of joint inflammation and associated with mechanical injury in patients with OA, the release of inflammatory mediators such as cytokines37 can induce a peripheral sensitisation and lead to a decrease in the excitation threshold (allodynia=normally innocuous stimuli may evoke a pain response) or an increase in responsiveness to supra-threshold stimuli of peripheral nociceptors (hyperalgesia=noxious mechanical stimuli can evoke exaggerated response). Corticosteroids and NSAIDs act by inhibiting prostaglandins, which are proinflammatory mediators involved in peripheral sensitisation. Other than peripheral sensitisation there also is evidence that central sensitisation is involved in the pathogenesis of pain in OA.38 New therapeutic molecules can target the nociceptors to attenuate the peripheral sensitisation phenomenon, which characterises OA. Nerve grow factor (NGF) has been detected in the OA vascular channels at the osteochondral junction, and osteochondral angiogenesis is associated with increased NGF expression. This may facilitate the sensitisation or the invasion into the articular cartilage of sensory nerves.39 The expression of substance P and calcitonin gene-related peptide (CGRP) are increased by NGF. Different anti-NGF molecules are under investigation for the treatment of OA pain. The clinical trials with anti-NGF drugs, such as tanuzemab, have demonstrated a significant reduction in knee pain compared with the control population.40 ,41 Nevertheless, in 2010 the Food and Drug Administration (FDA) suspended clinical trials because of cases of rapidly progressive OA in some patients. After reviewing of all the cases associated with anti-NGF treatment and concomitant use of NSAIDs, in 2012 the FDA recommended reinitiating trials with an alert of avoiding coprescription of NSAIDs.42
CGRP is another potential and interesting molecule for treating OA pain. CGRP acts through the calcitonin receptor-like receptor (CLRL) and the cofactor RAMP1. There is evidence indicating an association between joint levels of CGRP and pain during OA.43 ,44 CGRP has been found colocalised with NGF in the vascular channels at the osteochondral junction in OA,39 and it seems to be implicated in peripheral sensitisation in experimental OA.45 Moreover, CGRP is expressed in the meniscus46 and synovium47 and its expression correlates with chondral degeneration. Small molecule CGRP receptor antagonists have been recently developed and their efficacy in treating migraine pain in human subjects and OA pain in the monosodium iodoacetate animal model has been demonstrated.48 These findings indicate that CGRP represents an important neuropeptide that can be targeted in the treatment of OA pain. The presence of pain has been shown to correlate with MRI findings of moderate or large effusions, as well synovial thickening.49–51 In addition, pain has been demonstrated to correlate with synovitis in patients undergoing meniscectomy without radiological signs of OA.52 OA pain has been associated with bone marrow lesions detected on MRI and changes in bone marrow oedema have been correlated with fluctuation in OA knee pain.50 ,53 According to these observations, targeting subchondral bone and synovial inflammation may be reasonable therapeutic targets to address pain pathogenesis in OA, and this will be discussed in the next paragraphs.
Targeting subchondral bone
Bone marrow lesions identified by MRI in patients with OA are usually poorly circumscribed areas seen on fat suppressed images that occur in weight-bearing areas just underneath the cortical bone. Histologically, they are characterised by the presence of microfractures, increased osteoclasts resorption and woven bone.54 Bisphosphonates are antiresorptive agents that mainly act on osteoclasts. The major rationale for using these drugs to treat OA pain is related to the abnormal bone turnover in OA, which leads to a zone of osteoporosis beneath the subchondral plate, which results in decreased bone mechanical properties predisposing to bone microfractures.8 Osteoclasts contribute to vascular channel formation at the osteochondral junction resulting in vascular and neural invasion of the calcified articular cartilage and exposing nerves to proinflammatory mediators from the synovial fluid.39 Furthermore, osteoclasts may reduce PH at the osteochondral junction resulting in a sensitisation of the sensory nerves.55 A recent published review of the use of bisphosphonates for management of OA pain concludes that there is limited evidence that biphosphonates are effective in treatment of OA pain.56 Nevertheless, some limitations such as different duration of the treatment, dose, route of administration, lack of long-term data on OA joint structure modification have been highlighted.56 The efficacy of bisphosphonates in OA is controversial and some authors hypothesize that only the osteoporotic subchondral bone phenotype may benefit from antiresorptive treatment by decreasing bone remodelling and reducing bone resorption.15 ,57 Lack of efficacy of bisphosphonates may be explained by the limited potency of some molecules and by the timing of treatment in a heterogeneous diseases with different phases. Moreover, structural disease modification is measured in OA mainly by joint space narrowing, which is influenced by joint tissues that may not be a source of pain. In the literature, pain in OA has been associated with bone marrow lesions and synovitis. Interestingly, a recent study has shown that Zoledronate, a potent bisphosphonate, significantly reduces pain and bone marrow lesions at 6 months in patients with OA and compared to controls.58 This study highlights the concept of using the right treatment at the right time. Subcutaneous tiludronate has been showed to have an effect in a OA model of dogs in reducing not only pain and increasing subchondral thickness but also synovitis.59 This data are consistent with the effect of bisphosphonates on subchondral bone turnover but also suggests a possible action on synovitis.60
Strontium ranelate has recently demonstrated to have a structure modifying effect (evaluated semiautomatically by joint space narrowing) at both 1 and 2 g doses and a beneficial effect on pain at 2 g/day (but not with 1 g/day) compared with placebo over 3 years of treatment.61 ,62 Importantly, strontium ranelate has been shown to reduce cartilage volume loss in the tibial plateau and bone marrow lesion progression in the medial knee compartment as assessed by MRI.63 Its further development is hampered by side effects, such as cardiovascular events.64 Nevertheless, the above described SEKOIA study61 ,62 is a landmark study, since it showed that the reduction of radiological joint damage was possible with the use of an antiosteoporotic drug.
Targeting inflammation
Unlike in RA, it has been demonstrated that blocking IL-1β in OA synovium did not result in inhibiting production of tumour necrosis factor α (TNF-α). In addition, treatment of osteoarthritic synovium with anti-TNFα agents did not block IL-1β.65 Anti-TNF-α has not been shown to be effective in preventing the erosive structural damage of patients affected by erosive hand OA;66 while 70% of patients with knee OA (N=20) achieved an OARSI/OMERACT response in an open-label evaluation of adalimumab over 12 weeks.67 Blocking iNOS, which is a proinflammatory mediator in OA, failed to slow down or decrease OA progression in a 2-year randomised, double-blind, placebo-controlled, multicentre study of an oral selective iNOS inhibitor in patients with symptomatic OA of the knee.68 Even treatments with a single intra-articular injection of IL-1 receptor antagonist (IL-1Ra)69 and subcutaneous injections every 4 weeks for 3 months with an anti-IL-1 RI monoclonal antibody, which inhibits both IL-1α and IL-1β, has not shown a difference in terms of WOMAC scores in treated patients compared with placebo in knee OA.70 Interestingly, preoperative intra-articular injection of IL-1Ra significantly reduced pain and synovial fluid cytokine levels in a small study (N=11) in patients undergoing ACL reconstruction for ACL rupture.71 ,72 Since ACL rupture has been correlated with the development of OA, this data suggests that anti-IL-1β may be effective in the early phases of the disease, highlighting the concept that is important to identify distinct subsets of patients for treatment interventions.
Bone involvement in RA
The physiological bone remodelling cycle includes activation of osteoclasts from precursors, resorption, reversal phase and bone formation mediated by osteoblasts. This system permits individuals to adapt the skeleton to mechanical forces, to repair damage and contributes to calcium and phosphorus mineral ion homeostasis. During adulthood, prior to the arrival of menopause and the effects of aging, the resorptive and formation processes are intimately coupled in a state of equilibrium. In the inflammatory joint diseases this balance between bone catabolism and anabolism is lost. Understanding the physiology of this process provides insight for therapeutic interventions. The major mechanism of ‘coupling’ bone remodelling after the resorption phase includes the release of growth factors trapped in the bone matrix, which initiates the phase of bone formation. This provides a mechanism for quantitatively linking bone resorption and the initiation of bone formation.
Bone erosions in RA
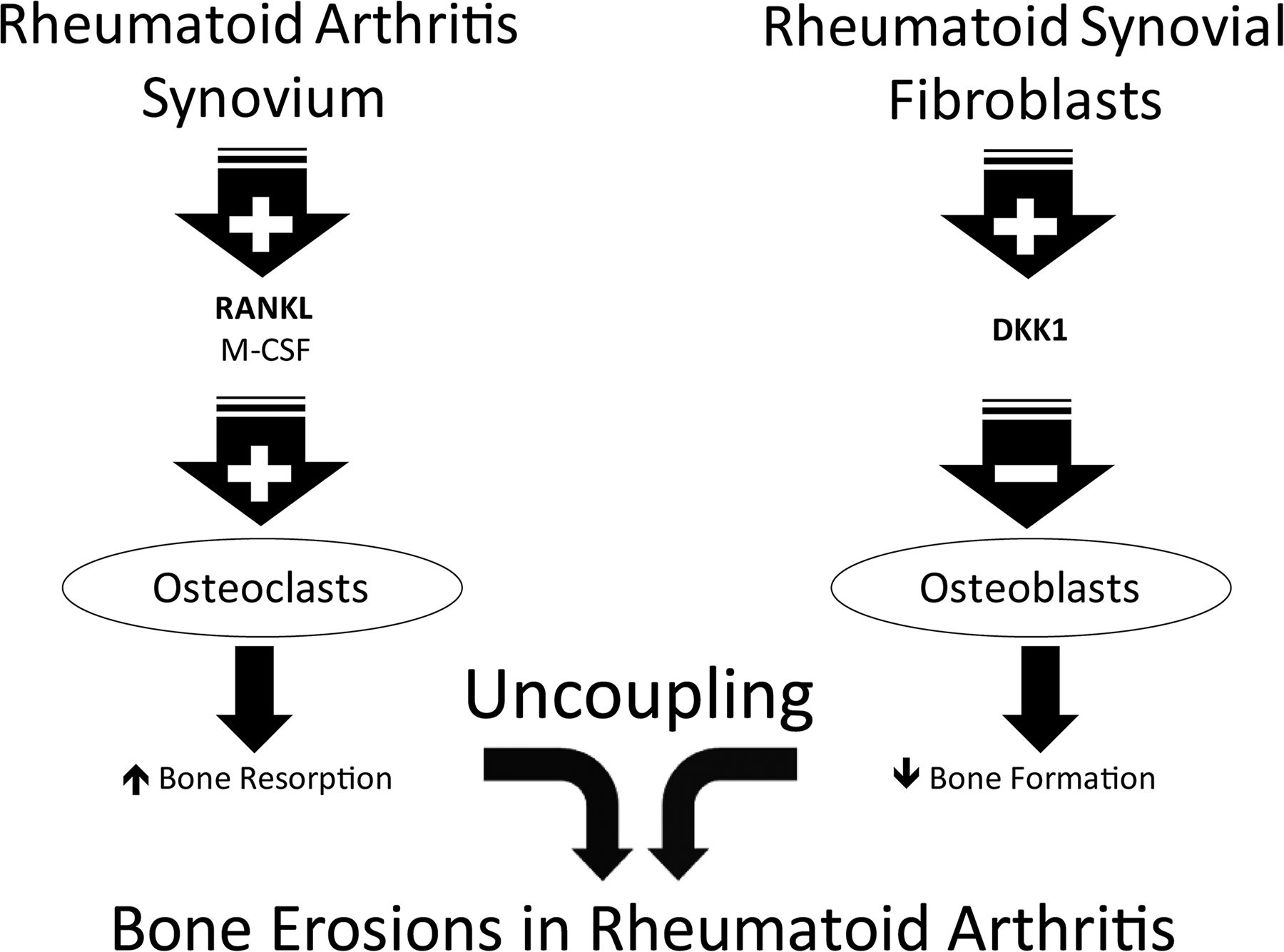
Osteoclasts are the essential cells that mediate the resorptive process under physiological conditions. In RA cells with phenotypic features of osteoclasts are present in resorption lacunae at the bone synovial interface.73 The pannus and inflammatory synovium are a very rich source of myeloid precursors and immunomodulatory and proinflammatory factors with osteoclastogenic activity.74–78 Indeed, macrophage lineage cells can differentiate into the osteoclasts, induced by factors produced by RA synovium, in particular RANKL and macrophage colony-stimulating factor (M-CSF). Of interest, the synovium adjacent to the resorption sites has been shown to be an abundant source of RANKL.79 It has been demonstrated that blocking osteoclasts represents a rational approach to prevent bone resorption in RA.80 RANKL knock-out (KO) mice lack osteoclasts and develop a severe form of osteopetrosis. To define the role of osteoclasts in the pathogenesis of bone erosions, serum from arthritic K/BxN mice containing pathogenic antiglucose-6-phosphate isomerase antibodies was transferred to RANKL gene KO mouse resulting in the development of arthritis that histologically resembles RA. In the absence of osteoclasts, despite marked synovial inflammation and pannus formation, the RANKL KO mice do not develop significant bone erosions compared to control mice.80 Similar results have been obtained in two other RA animal models lacking osteoclasts.81 ,82 This highlights the concept that osteoclasts play an essential role in the pathogenesis of focal bone erosions in RA.
In RA in addition to an increased bone resorption there is a decrease of bone formation at the erosion sites in active RA, indicating the presence of uncoupling of bone remodelling. Of interest when the joint inflammation is reduced by treatment there is partial restoration of bone formation.83
Regulation of osteoblast differentiation involves both Wnt ligands and BMPs as inducers of bone formation. Sclerostin and DKK-1 are inhibitors of the classical canonical Wnt/β-Catenin signaling pathway and they block the interactions between the receptor complex consisting of LRP5/6 and frizzled and Wnt resulting in a degradation of β-catenin and inhibition of bone formation.84 Antisclerostin antibodies have been demonstrated to increase bone mass in animal models of osteoporosis and fracture repair.85–87 Rheumatoid synovial fibroblasts are a very rich source of DKK188 and TNF-α is an important inducer of DKK-1. At least in part, the uncoupling of bone formation and the lack of bone repair in the rheumatoid process could be attributed to the production in the synovial tissue of this inhibitor of the Wnt pathway. Mechanisms leading to bone erosions in RA are summarised in figure 2.
{kind=link}
{kind=link}
Uncoupled bone remodelling and bone erosion formation in rheumatoid arthritis. In rheumatoid arthritis patients an increased bone resorption and a decreased bone formation lead to bone erosion formation.
Systemic bone loss in RA
In the pathogenesis of RA, two phases have been recognised: (1) an autoimmune phase, induced by environmental factors such as smoking, acting in a certain genetic background that imparts susceptibility. During this autoimmune phase, patients may develop rheumatoid factor (RF) and anticitrullinated protein antibodies (ACPA) over 10 years before the clinical manifestations of the disease.89–91 (2) an inflammatory phase, possibly induced by an infection or other environmental triggers, which leads to a chronic inflammatory state, which can ultimately affect multiple organs, including the bone.92 Both phases can induce bone alterations. Bone is one of the key organ targets in RA. In the literature there are many examples of an increased fracture, including vertebral and non-vertebral and, hip. Fracture risk in patients with RA is due to not only to trabecular but also cortical bone loss.93–98
Bone structure alterations, including reduced bone volume and bone mineral density (BMD), cortical bone thickness and increased fenestration have been seen in ACPA positive non-arthritic individuals compared to ACPA-negative controls.99 There are several observations supporting these data and demonstrating that bone metabolism is altered before the onset of the disease.100–102 Moreover, ACPA-positive patients are at risk for developing severe bone erosions with extensive resorption of the trabecular network. ACPAs isolated from sera of patients with RA have been shown to induce osteoclastogenesis and bone resorption in vitro.103 In addition, ACPA injections in immunodeficiency mice induce bone loss after 4 weeks due to increased osteoclast formation.104 ACPAs can induce osteoclast activation and bone loss starting 10 years before the onset of the clinical phase of the disease, prior to a second hit, which is associated with the appearance of overt synovitis.99 It has been suggested that the preclinical phase of RA primes the joint to the inflammatory phase because cortical fenestration induced by osteoclastic activity lead to a communication between bone marrow and synovial tissue. Bone marrow oedema in subchondral bone in RA predicts the development of bone erosions and there is evidence suggesting than plasma cells can trigger osteoclastogenesis by local production of antibodies.105 The synovial inflammation induces bone loss triggering an imbalance between bone resorption and formation similar to what occurs with some tumours.106 In an inflamed joint, the resorptive process immediately starts with the onset of the disease. Osteoclast precursors and osteoclasts has been found respectively after 2 and 5 days in the collagen-induced arthritis mouse model.107 The synovial inflammatory tissue instruct T cells to produce M-CSF and RANKL which induce osteoclastogenesis.108
New treatments targeting bone in RA
RANKL is highly expressed in the synovial membrane of patients with RA leading to osteoclast differentiation and activation.109 ,110 Treatment with denosumab, a human RANKL monoclonal antibody, increases lumbar spine BMDand reduces vertebral and non-vertebral fracture rates in postmenopausal women with osteoporosis,111 but also protects patients with RA from bone erosions, by arresting osteoclast formation and activation.112 ,113 Nevertheless denosumab does not have an effect on inflammation and its use would need to be combined with an anti-inflammatory strategy. Many cytokines including TNF-α, IL-1β, IL-6 are involved in the osteoclast activation process leading to bone loss in patients with RA.114 It has been demonstrated that TNF-α is a very potent inducer of osteoclasts.115 ,116 So the inhibitions of these cytokines can block bone erosions in addition to inhibitory effects on inflammation. IL-17 is one of the major drivers of RANKL production by synovial fibroblasts and is a potential target for the treatment of inflammatory arthritis and associated bone disease. As discussed above, TNF-α is able to block bone formation by production of inhibitors of osteoblast differentiation such as Dkk1 and sclerostin, in addition to its ability to enhance osteoclast differentiation.88 ,117 ,118
Bone involvement in SpAs
The SpAs are a heterogeneous group of chronic inflammatory diseases of the skeleton and associated soft tissues, including ankylosing spondylitis (AS), psoriasic arthritis (PsA), inflammatory bowel disease-associated arthritis (IBD), reactive arthritis, juvenile SpA and undifferentiated SpA. All subtypes of SpA are characterised by inflammation and structural damage of the axial and/or peripheral skeleton. The inflammatory process presents as osteitis, enthesitis, synovitis and may produce joint destruction with loss of articular cartilage and bone erosion. On the other hand, and in contrast to RA, skeletal abnormalities are manifest as new bone formation leading to bony fusion of the sacroiliac joints, syndesmophyte formation (with bridging of the intervertebral spaces) and enthesophyte formation. These processes, that are the hallmarks of the SpAs, contribute significantly to permanent disability and loss of function of the patients. Skeletal growth during development and growth, occurs by two different mechanisms: endochondral bone formation, by which the skeletal elements are defined by an intermediate cartilage stage; and membranous bone formation, that results from direct new bone formation. These processes rely on a number of molecular signaling pathways, that include bone morphogenic proteins (BMPs), Wnts, hedgehog, fibroblast growth factor, notch and parathyroid hormone-like peptide signaling. Although relatively few data are available for SpAs, animal models and human data suggests that both endochondral and intramembranous bone formation contribute to ankylosis in patients with SpA.119–121
A number of well-designed studies in animal models have highlighted the role of BMP and Wnt signaling in the process of ankylosis in SpA patients. Lories et al120 studied the spontaneous arthritis model in aging male DBA/1 mice to better understand the molecular mechanism of ankylosing enthesitis, and demonstrated that different BMPs are expressed during the process of ankylosis in this model.121 In the spontaneous ankylosing enthesitis model, the over-expression of noggin, a BMP antagonist, produced an inhibition of ankylosis.122 In another study, in the human TNF transgenic mouse model of arthritis, Diarra et al88 ,123 established the link between Wnt signaling and new bone formation. They studied the effect of blocking Dickkopf-1 (DKK1), an antagonist of Wnt signaling pathway. By blocking the DKK1, the phenotype of the disease shifted from joint destruction to joint remodelling with formation of osteophytes in the peripheral joints and ankylosis of the sacroiliac. The blockade of DKK1 function is associated with the activation of the canonical Wnt pathway and increased levels and nuclear translocation of β-catenin with resultant increased bone formation.
Human data further supported these animal observations. Analyses of pathological tissue from patients with SpA revealed the presence of BMPs and activation of BMP signaling in peripheral entheseal lesions.120 ,121 In addition, Diarra et al88 found that serum levels of DKK1 are very low or absent in patients with AS as compared to subjects with RA.
In conclusion, these and other experimental data strongly suggest that BMPs and Wnt signaling through β-catenin, play key roles in the pathogenesis of the enhanced periarticular bone formation and ankylosis in the SpAs. Current data, particularly in animal models, suggest that BMPs and Wnt pathways might represent potential targets for the prevention of ankylosis in SpA.
New treatments in SpAs
The introduction of anti-TNF drugs produced significant and relevant success in the management of SpAs. Anti-TNF therapies are highly effective in treatment of pain and stiffness in patients affected by SpAs, but these drugs have not been shown to slow the radiographic progression in clinical trials.124 However, the development of radiographic changes in SpAs is slow and placebo-controlled clinical trials have been carried out at a maximum for 24 weeks for ethical reasons. Interestingly, two recent observational studies in patients with AS, respectively at 8 years and up to 6.3 years showed a potential effect of anti-TNF therapy on slowing radiographic progression other than reducing inflammation.125 ,126
In addition, high-intensity or continuous NSAIDs have been shown to affect the AS radiographic progression, respectively, in a 2 year open-label randomised clinical trial and in a retrospective observational study. Interpretation of these results is limited by the fact that the first study had an unblinded design, while the second had a potential selection bias.
There are two emerging biologics, primarily developed in psoriasis and PsA, which seem to be effective not only in reducing inflammation but also in preventing bone formation in AS: ustekinumab (UST), a compound targeting IL-12/IL-23 via the p40 subunit of both cytokines and the anti-IL-17 antibody secukinumab (SEC). T cells differentiated to the Th17 phenotype under the influence of IL-23 are characterised by the production of IL-17. IL-23 and IL-17 levels are increased in blood samples of patients with AS and detected in synovial biopsies of these patients. Moreover, IL-23 receptor polymorphisms are associated with susceptibility to SpAs.127 The primary structure involved in spondyloarthropathy is the enthesis, and IL-23 has been demonstrated to stimulate enthesitis and periosteal bone formation in a collagen antibody-induced animal model. Blocking IL-23 at the time of disease induction reduced clinical disease scores and enthesal inflammation in the mice.128 Scherlock et al identified an enthesal resident T-cell population that express the IL-23 receptor in this mice model and showed that entheseal tissues respond to IL-23 by increasing the expression of IL-17 and Il-22 and of BMP7, which they implicated in the enthesal ossification mechanisms.129
UST has been recently shown to demonstrate significant clinical improvement in 20 patients with AS in a prospective, proof of concept clinical trial.130
SEC showed, in a small pilot study (n=29), an ASAS 20 response at week 6 in 60% of patients with AS compared with 17% of placebo group.131 These data identify the IL-23/Th17 pathway as an important key target in SpAs.
Conclusions
Several aspects of bone involvement in RA, SpAs and OA have been highlighted during the last two OsteoRheumatology meetings, held in 2013 and 2014. Although, a number of issues still remain to be clarified, it seems quite clear that in clinical practice, as well as in basic and translational research, there is a need for more knowledge of the interactions between the cartilage, the immune system and the bone. In this context, ‘OsteoRheumatology’ represents a new discipline that provides a greater insight into this interplay, in order to face the multifactorial and complex issues underlying common and disabling rheumatic diseases.
References
Footnotes
Contributors MF, AG, GB and GS took part in review, conception and design. MF and AG participated in drafting the manuscript. MF, AG, PG, SRG, WL, GS and GB critically reviewed the manuscript.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.