Article Text

Abstract
Objectives Suramin is an old drug used for the treatment of African sleeping sickness. We investigated therapeutic repositioning of suramin to protect against cartilage damage, as suramin may interact with tissue inhibitor of metalloproteinase-3 (TIMP3).
Methods In vitro extracellular matrix (ECM) accumulation and turnover in the presence or absence of suramin were studied in the ATDC5 micromass model of chondrogenesis and in pellet cultures of human articular chondrocytes from osteoarthritis and control patients, by gene expression, protein analysis, colorimetric staining, immunoprecipitation, fluorimetric analysis and immunohistochemistry. To study suramin in vivo, the drug was injected intra-articularly in the papain model of joint damage. Disease severity was analysed by histology, immunohistochemistry and contrast-enhanced nanofocus CT.
Results In ATDC5 micromasses, suramin increased TIMP3 levels and decreased the activity of matrix metalloproteinases (MMPs) and aggrecanases. Suramin treatment resulted in increased glycosaminoglycans. This effect on the ECM was blocked by an anti-TIMP3 antibody. Direct interaction between suramin and endogenous TIMP3 was demonstrated in immunoprecipitates. Mice treated intra-articularly with suramin injections showed reduced cartilage damage compared with controls, with increased TIMP3 and decreased MMP and aggrecanase activity. Translational validation in human chondrocytes confirmed increased TIMP3 function and reduced cartilage breakdown after suramin treatment.
Conclusion Suramin prevented loss of articular cartilage in a mouse model of cartilage damage. The effects appear to be mediated by a functional increase of TIMP3 and a subsequent decrease in the activity of catabolic enzymes. Thus, suramin repositioning could be considered to prevent progressive cartilage damage and avoid evolution toward osteoarthritis.
- osteoarthritis
- chondrocytes
- treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
KEY MESSAGES
What is already known about this subject?
Tissue destructive enzymes such as matrix metalloproteinases and ADAMTs enzymes contribute to cartilage loss after injury and progression toward osteoarthritis.
Increasing the levels of TIMP3, an endogenous inhibitor of these enzymes, is considered a therapeutic target.
What does this study add?
Suramin, an old drug used for the treatment of sleeping sickness, can bind to TIMP3 and increase its levels in the extracellular matrix.
This results in effective cartilage protection in an in vivo mouse model of cartilage injury.
How might this impact on clinical practice?
Suramin or suramin analogues may be considered for further clinical development to protect the articular cartilage.
Introduction
Cartilage damage can be caused by high-impact trauma and trigger a detrimental cascade eventually leading to osteoarthritis, the most common chronic and debilitating joint disease.1 2 Interventions to prevent disease progression from joint injury toward osteoarthritis are a large unmet medical need. Tissue destructive enzymes such as matrix metalloproteinases (MMPs) including different collagenases and such as aggrecanases ‘a disintegrin and metalloproteinase with thrombospondin motifs’ 4 (ADAMTS4) and 5 play a major role after acute joint injury and in the development of osteoarthritis.3 4 Indeed, these proteases break down collagens and proteoglycans in the extracellular matrix (ECM) of the articular cartilage.5 Thus, tissue destructive enzymes represent interesting targets for therapy, but specificity and toxicity issues have hampered the development of pharmacological inhibitors.6–8 Therefore, deciphering alternative ways of controlling pathological proteolysis in cartilage could result in significant therapeutic progress.2 9 10
De novo development of drugs is a time-consuming and costly process, which is often inefficient. Drug repositioning is an emerging and attractive mode of therapeutic discovery for the application of known drugs to treat new indications.11 12 This pharmaceutical strategy substantially reduces the cost and duration of the drug development pipeline and also the risk of unforeseen adverse events. We explored a chondroprotective role for suramin, a 100-year-old drug, also known as Germanin or Bayer-205, and used for the treatment of African sleeping sickness caused by the trypanosome parasite.13 Drug repositioning for suramin has included tests for diseases such as cancer,14 15 autism,16–18 as well as multiple viral infections such as HIV,19 Ebola,20 dengue21 and rift valley fever,22 although with limited success until now.23 However, the use of suramin has not yet been explored in the context of cartilage damage and osteoarthritis.
Suramin is a polysulfonated naphthylureic molecule that is negatively charged and has the capacity to bind with basic side chains of proteins, thereby potentially affecting their biological activities.24 Interestingly, suramin has been used in vitro to isolate TIMP3 from rat uterine protein extracts.25 TIMP3 is a major inhibitor of several MMPs and aggrecanases, in particular pro-MMP2, MMP3, MMP9, MMP13, ADAMTS4 and ADAMTS5.26 Parallel and independent to our study, Chanalaris et al demonstrated that suramin can prevent ex vivo cartilage degradation by increasing TIMP3 levels and preventing TIMP3 endocytosis.27 In addition, suramin could also inhibit purinergic receptors,28 has been linked with inhibition of Wnt signalling29 and can interact with and inhibit fibroblast growth factor-2.30 Based on these different characteristics, we hypothesised that suramin may have cartilage-protective properties.
Materials and methods
ATDC5 micromass cultures
ATDC5 cells were cultured in growth medium (1:1 Dulbecco’s modified Eagle’s medium (DMEM): Ham’s F-12 mix (Gibco)) containing 1% (vol/vol) antibiotic–antimycotic (Gibco), 5% foetal bovine serum (FBS) (Gibco), 10 µg/mL human transferrin (Sigma) and 30 nM sodium selenite (Sigma). Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
High-density micromass cultures of ATDC5 cells were grown to study chondrogenic differentiation. Cells were trypsinised, washed and resuspended at 2×107 cells/mL in a chondrogenic medium made of DMEM-F12 enriched by 1% (vol/vol) antibiotic–antimycotic, 5% FBS, 5 µg/mL human transferrin and 1× of ITS (Insuline , Transferin, Selenite) premix (resulting in 10 µg/mL insulin, 5 µg/mL human transferrin and 30 nM sodium selenite) (Life Technologies). One droplet (10 µL) was carefully placed in the centre of each well of a 24-well plate. Cells were allowed to adhere for 2 hours at 37°C, followed by addition of 500 µL chondrogenic medium supplemented with or without suramin 10 µM (Sigma). After 14 days, induction of hypertrophic differentiation and mineralisation were induced by the mineralisation medium made of α-minimum essential medium Eagle (Gibco) containing 1% (vol/vol) antibiotic–antimycotic, 5% FBS, 5 µg/mL human transferrin, 1× of ITS premix, 50 µg/mL ascorbic acid-2-phosphate (Sigma) and 7 mM β-glycerophosphate (Sigma). Micromasses and supernatants were collected at time points 1, 7, 14 and 21 days. Each time point was processed with three technical replicates.
To neutralise tissue inhibitor of metalloproteinase (TIMP) 3 activity, micromasses were treated at day 1 with 10 µg of anti-TIMP3 (Abcam ab39184) or 10 µg of isotype control IgG (Abcam ab199376). Antibody treatments were renewed every 3 days. Plates were collected at days 7 and 14. Each time point was processed with three technical replicates.
Human articular chondrocyte pellet cultures
Primary human articular chondrocytes were studied in pellet cultures. The human articular chondrocytes were isolated from the hips of patients undergoing total hip replacement surgery. Healthy articular chondrocytes were obtained from patients undergoing hip replacement for osteoporotic or malignancy-associated fractures. The University Hospitals Leuven Ethics Committee and Biobank Committee approved the study, and specimens were taken with patients’ informed consent. Chondrocytes were isolated after cutting cartilage slices into pieces of 4×4 mm. Samples were washed three times in 1% (vol/vol) antibiotic–antimycotic/phosphate-buffered saline (PBS) and incubated with 1 mg/mL pronase (Roche)/DMEM-F12 at 37°C for 30 min at slow rotation 100 rpm followed by an overnight incubation with 1 mg/mL collagenase B (Roche)/DMEM-F12 at 37°C. Cells were filtered through a 70 µm cell strainer (Corning), washed twice with PBS, seeded at 1×106 cells/T75 flask and cultured for 7–14 days in maintenance medium (DMEM-F12, containing 1% (vol/vol) antibiotic–antimycotic (Gibco), 10% FBS (Gibco) and 5% L-glutamine (Thermo scientific)). Passage 1 cells were used for experiments.
Pellet cultures were obtained by trypsinising, washing and resuspending human articular chondrocytes at 2×106 cells/mL in differentiation medium (DMEM-F12 enriched by 1% (vol/vol) antibiotic–antimycotic, 10% FBS, 1× of ITS premix (resulting in 10 µg/mL insulin, 5 µg/mL human transferrin and 30 nM sodium selenite) (Life Technologies), 50 µg/mL ascorbic acid-2-phosphate (Sigma) and 5% L-glutamine (Life Technologies)). One hundred microlitres of cell suspension was placed in 96 V well shaped plate (Greiner Bio-one) and rotated at 500 g for 10 min at 20°C. Cell pellets were collected at 1, 7 and 14 days for di-methylmethylene blue (DMMB) assay or treated with 4% paraformaldehyde (PFA) overnight for histological analysis and immunohistochemistry. Five-micrometre paraffin sections were stained with Safranin O–fast green.
Papain model of cartilage damage
The KU Leuven Ethical Committee for Animal Research approved all animal experiments. Osteoarthritis was enzymatically induced in 8-week-old male wild-type C57/Bl6 mice by intra-articular injection of a mixture of 2% papain (5 µL) (Sigma) and its activator 0.03 M cysteine (5 µL) (Sigma) into the right knee joints on the first day as described before.31–33 Control group received papain only, and the treated group received papain in combination with 0.1 mg suramin (Sigma). An equal volume of sterile PBS was injected into the left knee that served as a control knee. At day 3, mice received a second intra-articular injection of 0.1 mg suramin or PBS. Mice were sacrificed after 6 days, and hind limbs were isolated. These hind limbs were fixed with 4% paraformaldehyde (Millipore) overnight at 4°C. Whole knees were decalcified in 0.5 M EDTA (pH 7.5) for 15 days at 4°C and paraffin embedded to section at 5 µm in frontal plane for histology and TIMP3, VDIPEN and NITEGE immunohistochemistry. Samples were stained with Safranin O and haematoxylin for histology analysis. Articular cartilage damage was quantified by one blinded reader according to the OARSI (Osteoarthritis Research Society International) scoring system.34 Two independent randomised experiments were performed and consisted a total of 30 mice.
Contrast-enhanced nanofocus CT (CE-nanoCT)
Fixed mouse knee joints, dislocated to expose the femoral and tibial articular cartilage, as well as reference samples were stored in PBS at 4°C. All samples were transferred to a 20% Hexabrix 320 (Guerbet Nederland BV) solution in PBS for overnight incubation and were then wrapped into parafilm prior to scanning as described.35 The equilibrium contrast agent Hexabrix 320, a negatively charged ioxaglate, is repelled by anionic sulfated glycosaminoglycans (sGAG) resulting in an inversed staining of the sGAG content.
The applied nanoCT system was a nanoTom M (GE Measurement and Control Solutions, Germany). All acquisitions were performed using diamond-coated tungsten target. A 0.2 mm aluminium filter was used to reduce beam hardening. Mice knee scans were performed at 60 kV and 140 µA with a 500 ms exposure time, and an averaging of 1 and a skip of 1 (‘fast scan mode’) were chosen. A total of 2400 images were acquired over 360°, resulting in a scan time of 20 min. The isotropic voxel size of mouse knee scans was 2 µm, respectively. Reconstruction was performed using Phoenix datos|x CT software. Three-dimensional (3D) visualisation of the articular cartilage and bone of the mouse femora and/or tibiae mimics (Materialise, Haasrode, Belgium) was applied as described.35 To quantify the effective thickness of the articular cartilage, the cartilage region was selected using CTAn software (Bruker micro-CT, Kontich, Belgium) by manually drawing a region of interest (ROI) in every 20th cross-section throughout the joint surface. Delineating the total cartilage area was performed by readers, blinded for the experimental conditions and was based on the difference in grey scale from the surrounding (ie, hexabrix) and the structure to the subchondral bone. Selected ROIs were segmented and analysed in 3D; the effective thickness was calculated based on the volume and area surface data.
RNA extraction, complementary DNA (cDNA) synthesis and quantitative real-time PCR (qRT-PCR)
Total RNA from micromasses was isolated using the Nucleospin RNA II kit (Macherey-Nagel) and reverse transcribed using the Revert Aid H Minus First Strand cDNA synthesis kit (Fermentas) following the manufacturers’ instructions. The maxima SYBR Green qPCR master mix (Fermentas) was used to assess the messenger RNA (mRNA) expression of aggrecan (Acan), Adamts4, collagen type 2a1 (Col2a1), collagen type 10a1 (Col10a1), Mmp9, Mmp13, TIMP3, Biglycan (Bcan), Decorin (Dcor) and Versican (Vcan) in the ATDC5 micromasses. Primer sequences are shown in the online Supplementary file 1. The following PCR conditions were used: 1 min at 95°C, 40 cycles of 15 s of denaturation at 95°C, followed by 45 s of annealing/elongation at 60°C. Melting curve analysis was performed to determine the amplification of the specific product. Results were expressed using the comparative threshold method and were normalised to housekeeping gene S29 (ribosomal protein S29) mRNA level. The Rotor-gene 6000 detection system (Corbett Research, Westburg, Leusden, The Netherlands) was used for qRT-PCR measurements.
Supplementary file 1
Matrix composition analysis
ATDC5 micromasses were washed with PBS and fixed with 95% ice-cold methanol for 30 min at 4°C for staining. After washing three times with water, micromasses were either paraffin-embedded for immunohistochemistry or stained with Alcian Blue (AB) (0.1% AB 8GX (Sigma)) or Alizarin Red (AR) (1% AR (Sigma) in water pH 4.2), washed three times with water and air dried. Quantification of the staining was performed by dissolving the micromasses with 6M guanidine/HCl (Sigma) and by measuring the absorbance at 595 and 550 nm, respectively, with a spectrophotometer (BioTek Synergy).
Biochemical analysis of DNA and glycosaminoglycan (GAG) amount: DMMB assay
Harvested micromasses and pellets were digested at 56°C overnight with 0.1 M proteinase K in PBE buffer (1 mM EDTA, 10 mM Tris, pH 6.5). DNA amount was measured using the nanodrop (nanodrop 2000). GAG amount was measured at 525 nm with a spectrophotometer (BioTek Synergy) using DMMB dye added to 30 µL of pellet digestion or undiluted supernatant and bovine chondroitin sulfate (Sigma) as a standard.
Histology and immunohistochemistry
Paraffin-embedded micromasses, mouse knees and human pellets were sectioned at a thickness of 5 µm. Slides were deparafinised, quenched twice in 3% H2O2/PBS (Chem-Lab) for 10 min and blocked with 20% normal goat serum (Millipore) in TBS-0.1% Triton-X100 (TBS-Triton) (AppliChem) for 1 hour. Sections were incubated with primary TIMP3 antibody (Pierce, Thermo Scientific, PA5-26133) diluted 1:100, aggrecan antibody to C-terminal neo-epitope NITEGE (Pierce, Thermo Scientific, PA1-1746) diluted 1:100, aggrecan monoclonal antibody to N-terminal neo-epitope DIPEN (MD Biosciences, Zurich, 1042002) diluted 1:100 in 20% normal goat serum in TBS-Triton at 4°C overnight. This was followed by three washes in TBS-Triton and 1-hour incubation at room temperature with secondary antibody solution composed of peroxidase-conjugated goat anti-rabbit or mouse anti-rabbit secondary diluted 1:200 in 20% normal goat serum in TBS-Triton. Slides were rinsed and stained with diaaminobenzidine (DAB) chromagen (Dako) resulting in a brown staining and counterstained with haematoxylin (Sigma) for 30 s. Slides were washed with water and dehydrated with ethanol before mounting. Negative controls (normal rabbit IgG (R&D systems)) were included for all conditions. DAB quantification was performed with colour deconvolution plugin (Jacqui Ross, Auckland University) in ImageJ Software (NIH Image, National Institutes of Health, Bethesda, Maryland, USA).
Protein extraction and Western blot analysis
Proteins were isolated from the micromasses using cell extraction buffer (Thermo Scientific) supplemented with 1 mM phenylmethanesulfonyl (Sigma), 25 mM Na3VO4 (Sigma), 25 mM 10 µL NaF (Sigma) and 5% protease inhibitor cocktail (Sigma) using the Fastprep-24 tissue homogeniser (MP Biomedicals). A total of 20 µg of each lysate sample or 20 µL of supernatant sample or the pellet rest fraction representing the ECM was denatured for 5 min at 95°C, chilled on ice and separated on a 4%–12% Bis/Tris gel (Invitrogen) by electrophoresis using NuPage MES SDS Running buffer (Invitrogen). Then, proteins were transferred onto a polyvinylidine difluoride membrane (Millipore). After 2 hours in blocking buffer (TBS-0.1% Tween (TBST) supplemented with 5% non-fat dry milk) at room temperature, membranes were washed three times in TBST and incubated overnight at 4°C with primary antibody against TIMP3 (Abcam ab39184) at a 1:2000 dilution in blocking buffer. After three washes with TBST, each blot was incubated for 1 hour at room temperature with anti-rabbit conjugated with horseradish peroxidase (Jackson ImmunoResearch Laboratories) at a 1:5000 dilution in blocking buffer. Blots were visualised using SuperSignal West Femto Maximum Sensitivity Substrate system (Thermo Scientific) according to manufacturer’s recommendations. Images were acquired with the LAS-3000 mini CCD camera (Fujifilm). Densitometry analysis was performed with ImageJ Software (NIH Image, National Institutes of Health).
Fluorimetric spectroscopy
Changes in auto-fluorescent emission of suramin over the concentration range of 0–1000 µM were measured with a spectrophotometer (BioTek Synergy) at 25°C, using a 96-well, F bottom, chimney well, µ clear, black, Cellstar plate (Greiner). For the experiments to determine the interaction of suramin with TIMP3, the samples contained 10 µM suramin with or without 0.1 µg/mL recombinant TIMP3 (Sigma T1327). The samples were excited at 360 nm, and the emission spectrum was measured at 460 nm.
TIMP3 immunoprecipitation
Immunoprecipitation experiments were performed using the Pierce co-immunoprecipitation kit (Thermo Scientific). Columns were conditioned following the manufacturer’s recommendations. Antibody binding to the column was performed using 75 µg of either a mock antibody (donkey anti-goat IgG) as a control or C-terminal directed TIMP3 antibody (Abcam 187297). After antibody immobilisation, the columns were washed, and 100 µg of the rest fraction proteins was incubated overnight at 4°C under constant mixing. After four washings, retained proteins were eluted using 70 µL of elution buffer (Thermo Fisher, pH 3). Protein–suramin complexes were then detected by fluorimetric spectroscopy.
Enzymatic assay of papain
The papain activity was measured according to manufacturer’s enzymatic papain assay protocol. One unit papain will hydrolyse 1 M of Nα-benzoyl-L-arginine ethyl ester (BAEE) per minute at pH 6.2 at 25°C. In 10 mL reaction mix with final concentration of 56 mM BAEE (Sigma), 2.0 mM EDTA (Sigma), 5.0 mM L-cysteine (Sigma), 300 mM sodium chloride (Sigma) and 1.0 unit papain (Sigma), the pH of the reaction mix was monitored, and the time was recorded when the pH reached 6.2. The pH value of 6.2 was maintained by adding small volumes of 20 mM NaOH (Sigma), and the time was recorded when a total of 50 µl NaOH was consumed. This process was repeated for 10 min. Volume (in µL) of NaOH consumed was plotted versus the time (in seconds).
Statistics
GraphPad Prism was used to perform all analyses. One-way analysis of variance (ANOVA), followed by Tukey tests, or t-test was used for multiple and two-group comparisons. For experiments with multiple variables and/or repeated measurements, two-way ANOVA was used and included assessment of time or disease status–intervention interaction, time or disease status, and intervention. Normal distribution and equal variances were assessed, and where necessary, data were log transformed. Sidak test was used for multiple comparisons when interaction between time and intervention was significant. Data are shown as mean±SEM or as individual paired data points.
Results
Suramin treatment increases proteoglycans during chondrogenic differentiation
To study whether suramin augments ECM deposition in cartilage cells, we first used an in vitro model of chondrogenic differentiation and matrix production. ATDC5 cells are mouse chondrogenic precursor cells that differentiate toward chondrocytes when cultured in micromasses. To evaluate the levels of sulfated GAGs in the ECM of the micromasses, we performed AB staining and the DMMB assay. We observed a significant increase in proteoglycans in suramin-treated micromasses compared with controls (figure 1A–C). In contrast, mineralisation of the micromasses, assessed by AR staining, was not different between groups (online supplementary figure 1A, B). Remarkably, we detected no differences in mRNA expression levels of different matrix molecules (Acan, Col2a1, Col10a1, Bcan, Dcor, Vcan) (online supplementary figure 1C) and of tissue destructive enzymes (Mmp9, Mmp13 and Adamts4) (online supplementary figure 1C). Collectively, these results indicate that suramin has no direct effects on the transcription of these proteoglycans and tissue destructive enzymes and thus may likely increase proteoglycan content via post-transcriptional mechanisms.
Supplementary file 2

Suramin treatment increases proteoglycan accumulation during chondrogenic differentiation. (A) Alcian Blue staining of ATDC5 micromass cultures at day 1 (D1) and at day 21 (D21) showing increased proteoglycan content in the presence of suramin compared with controls, as quantified by (B) colourimetry at day 21 (*P<0.05, unpaired t-test), and by (C) di-methylmethylene blue (DMMB) assay determining total sulfated glycosaminoglycan content normalised to DNA content at different time points (P<0.001 for interaction between time and treatment, two-way analysis of variance; *P<0.001 between groups at days 14 and 21, Sidak multiple comparisons test). Data shown are representative of three independent biological replicates. Error bars indicate mean±SEM of three technical replicates per experiment.
Suramin increases the effects of TIMP3 and reduces MMP and aggrecanase activity in vitro
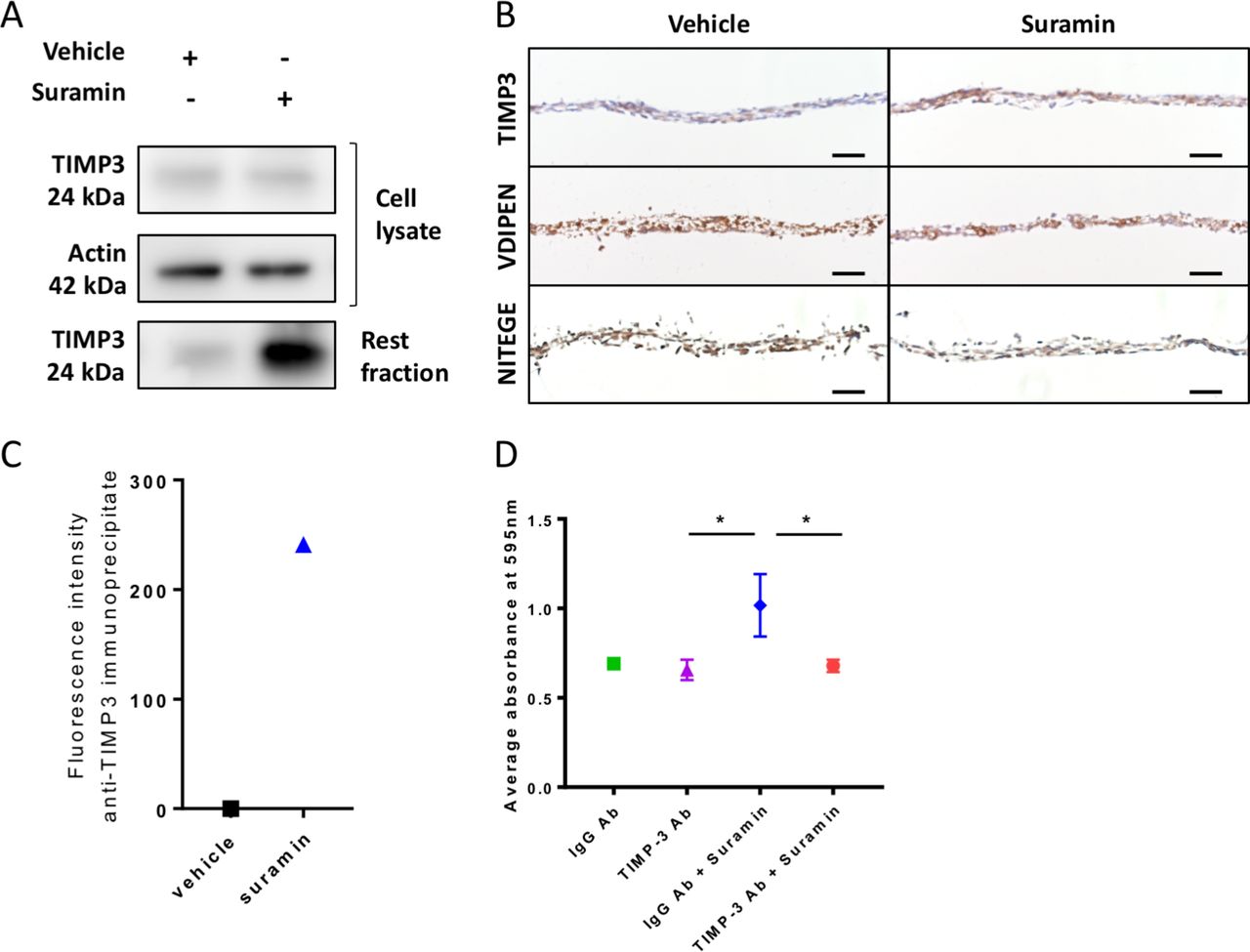
We next sought to decipher the mechanisms via which suramin increases ECM deposition in the chondrogenic differentiation assay. We hypothesised that suramin binds to TIMP3 and thereby increases the local concentration of the enzyme and its inhibitory effects in the ECM. To test our hypothesis, we measured TIMP3 levels and effects in our chondrogenic differentiation model. TIMP3 protein levels did not differ in the cell lysates but increased strongly in matrix extracts (rest fraction) from suramin-treated micromasses compared with controls (figure 2A). Increased TIMP3 levels in the suramin-treated micromasses were confirmed by immunohistochemistry (figure 2B). Furthermore, we detected reduced MMP and aggrecanase activity in suramin-treated micromasses as demonstrated by decreased staining of neo-epitopes VDIPEN and NITEGE, associated with MMP and aggrecanase activity, respectively (figure 2B). To assess a direct interaction between suramin and TIMP3, we took advantage of the fact that suramin exhibits a weak auto-fluorescent emission (near 400 nm), as shown in online supplementary figure 2A. On addition of recombinant TIMP3 to suramin, this fluorescence was considerably enhanced (supplementary figure 2B), indicating a direct interaction between the two compounds.36 Then, to prove that this interaction occurs with endogenous TIMP3 in suramin-treated micromasses, we performed immunoprecipitation using a specific anti-TIMP3 antibody. Effectively, we detected positive fluorescence in the immunoprecipitates from suramin-treated micromasses, while no signal was observed in vehicle-treated cells (figure 2C). Together, these data suggest that suramin increases proteoglycan content via augmenting TIMP3 levels and subsequently TIMP3’s inhibitory action on MMPs and aggrecanases. Thus, suramin may have protective effects against cartilage degradation.
Supplementary file 3
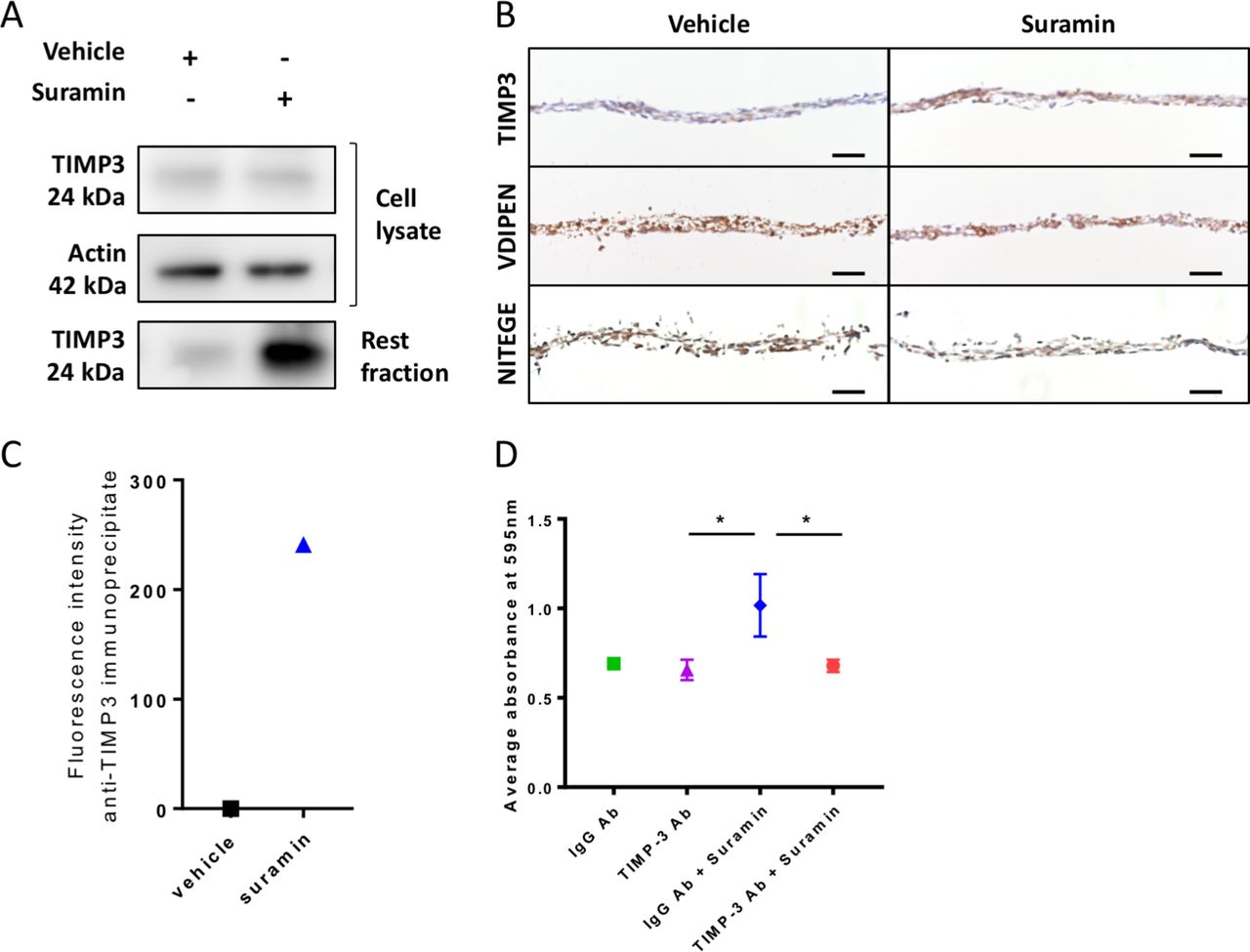
Suramin increases TIMP3 levels and reduces MMP and aggrecanase activity in vitro. (A) Western blot showing an increased amount of TIMP3 protein in extracellular matrix (rest fraction) of suramin-treated ATDC5 micromasses compared with controls after 21 days of differentiation culture. (B) Immunohistochemistry showing increased TIMP3 and decreased VDIPEN and NITEGE levels in suramin-treated micromasses compared with controls after 21 days of differentiation culture. Scale bar 50 µm. (C) Fluorescence intensity was measured on extracellular matrix proteins from micromasses (rest fraction) treated with suramin or vehicle, after anti-TIMP3 immunoprecipitation. Values are shown as the difference with the fluorescence emitted from anti-IgG immunoprecipitates. (D) Ten-micromole TIMP3 neutralising antibody blocks the effects of suramin in the ATDC5 micromasses at day 14; control is 10 µM IgG Ab. (*P<0.01, one-way analysis of variance followed by Tukey’s multiple comparisons test). Data shown are representative of three (A, B) or two (C) biological replicates. Error bars indicate mean±SEM of three technical replicates per experiment.
To determine whether the accumulation of TIMP3 is the main mechanism via which suramin increased matrix accumulation in micromasses, we neutralised TIMP3 in the micromasses using a specific antibody against TIMP3. Control IgG-treated cells showed increased proteoglycan accumulation after suramin treatment, whereas the anti-TIMP3 antibody completely blocked this effect of suramin (figure 2D). Thus, these results demonstrate that suramin promotes matrix deposition via a molecular mechanism involving TIMP3.
Suramin protects against cartilage damage in the papain-induced mouse model
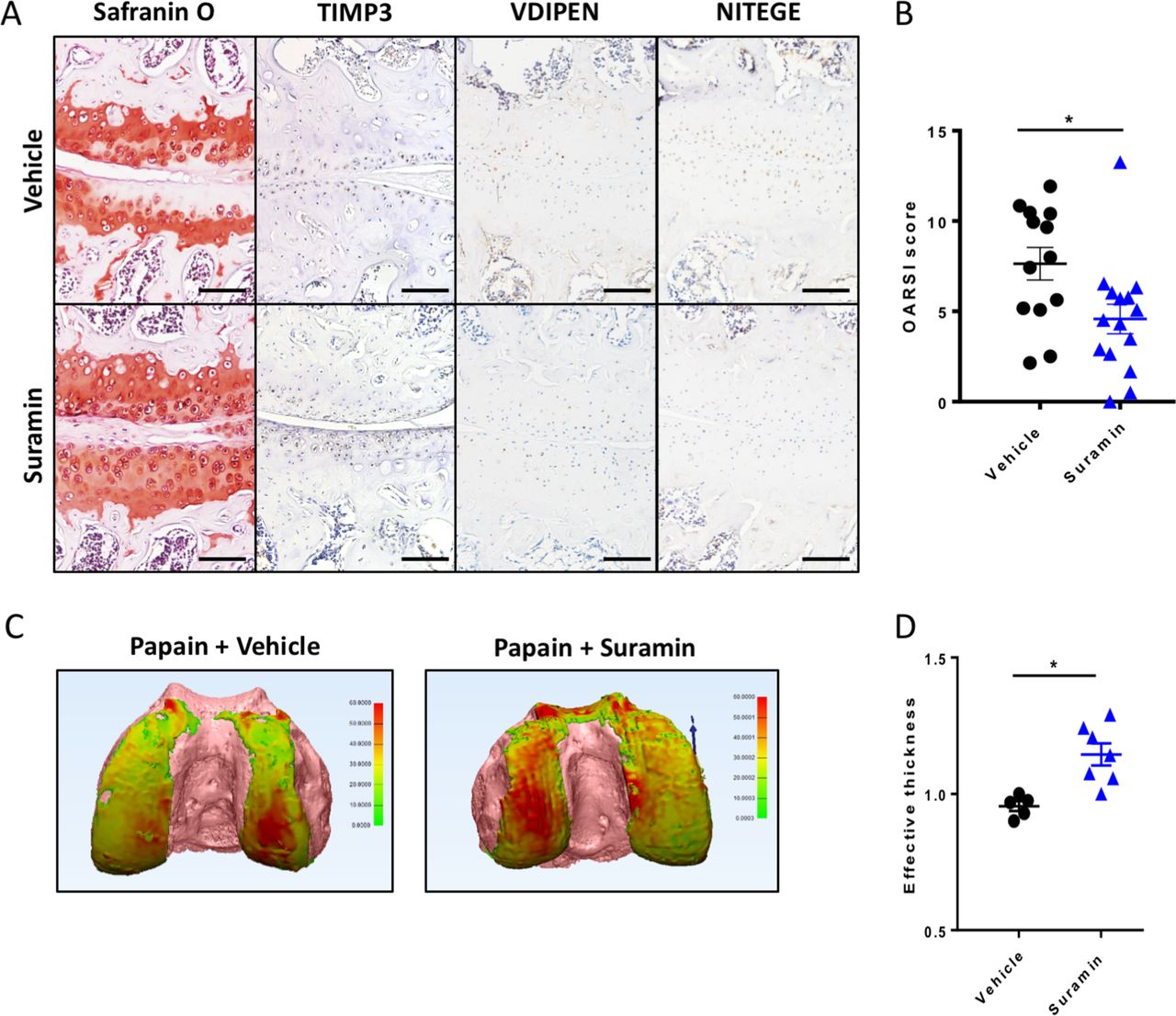
To assess the therapeutic efficacy of suramin in preventing cartilage damage, we used the papain-induced model of direct cartilage damage. In this model, 8-week-old mice are challenged by intra-articular papain injection. The papain enzyme cleaves proteoglycans in the cartilage, which triggers a cascade of cartilage breakdown. The enzymatic activity of papain remained intact in the presence of suramin (online supplementary figure 4). Suramin intra-articular co-injection significantly reduced the severity of cartilage loss in this model as shown by histology and disease severity scores (figure 3A, B). Suramin-treated knees showed increased levels of TIMP3 by immunohistochemistry (figure 3A). In addition, VDIPEN and NITEGE neo-epitopes were decreased in suramin-treated knees compared with controls (figure 3A). When we measured cartilage effective thickness by CE-nanoCT, we observed a significant difference in cartilage thickness in suramin-treated knees of mice exposed to papain, compared with vehicle-treated control knees after in vivo papain treatment (figure 3C, D). Thus, these in vivo data indicate that suramin reduces tissue damage after cartilage injury, by promoting a functional increase in TIMP3 in the joint.
Supplementary file 4
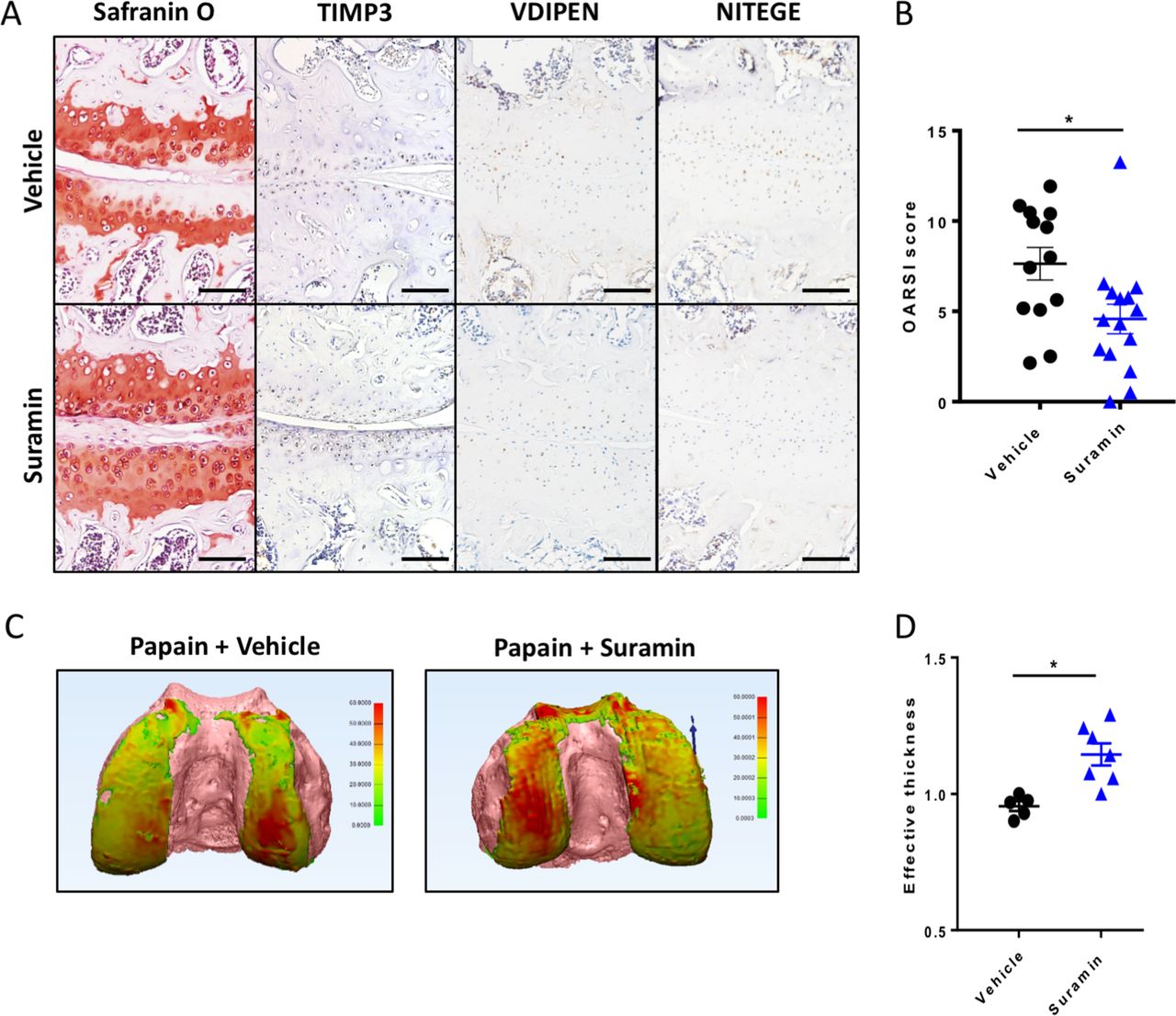
Suramin protects against osteoarthritis in the papain-induced mouse model. (A) Safranin O–eosin staining and immunohistochemistry of suramin and papain co-injected knees from in wild-type C57/Bl6 mouse showing cartilage preservation, increased TIMP3 and reduced VDIPEN and NITEGE levels compared with papain injection alone. Scale bars, 100 µm. (B) OARSI scoring of the medial knee compartment demonstrating that suramin treatment protects against papain-induced osteoarthritis (*P<0.001, unpaired t-test, n=13 and n=15). Data are obtained from two independent experiments. (C) Hexabrix contrast-enhanced nano-CT visualising cartilage in thickness maps on femoral bone (pink) from 9-week-old mice challenged with intra-articular papain with and without suramin treatment. (D) Medial femoral cartilage thickness quantification showing that suramin treatment reduces osteoarthritis development by increasing effective thickness of cartilage (normalised to left control knee; P<0.004 unpaired t-test, n=5–7). Data are obtained from a third in vivo experiment. Error bars indicate SEM.
Suramin increases TIMP3 levels and proteoglycan in human articular chondrocyte pellets
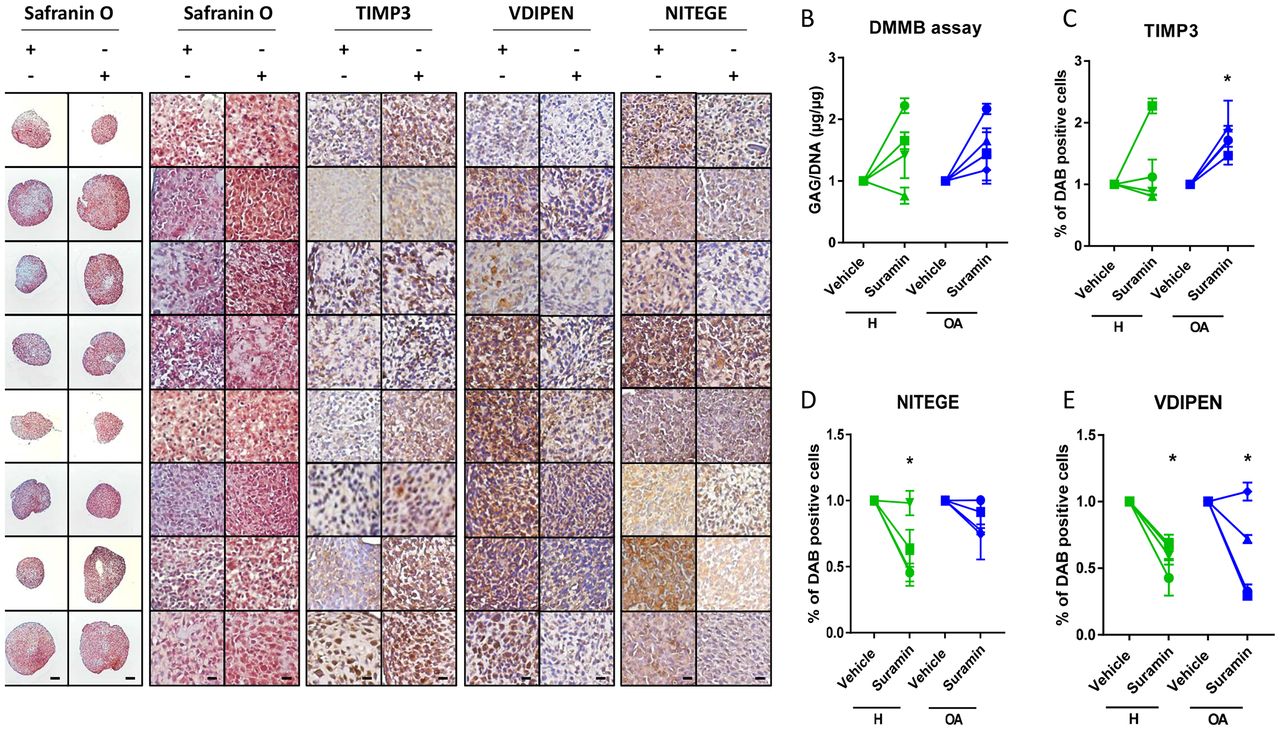
To investigate whether the protective effects of suramin and its associated molecular mechanism discovered in mouse cells and tissues are present in humans, we cultured primary human articular chondrocyte pellets derived from healthy and osteoarthritic patients and treated them with suramin. Suramin-treated pellets showed increased proteoglycan content compared with controls (figure 4A). GAG content increased in chondrocyte pellets treated with suramin compared with controls (figure 4B). TIMP3 levels were significantly upregulated in suramin-treated pellets, an effect that appeared specific for the osteoarthritic pellets (figure 4C). In addition, VDIPEN and NITEGE neo-epitopes were decreased after suramin treatment (figure 4D, E).
![[SP2.jpg]](https://rmdopen.bmj.com/content/rmdopen/3/2/e000604/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
![[SP3.jpg]](https://rmdopen.bmj.com/content/rmdopen/3/2/e000604/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
![[SP4.jpg]](https://rmdopen.bmj.com/content/rmdopen/3/2/e000604/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
{kind=link}
Suramin’s chondroprotective effects and molecular mechanism via TIMP3 are present in human articular chondrocytes. (A) Safranin O staining of human articular chondrocyte pellets demonstrating increased proteoglycan content after suramin treatment compared with controls. Scale bars 100 µm, right panels: higher magnification of Safranin O staining and immunohistochemistry on human articular chondrocyte pellets. Scale bars 20 µm. (B) Di-methylmethylene blue (DMMB) assay demonstrating increased sulfated glycosaminoglycan content in human articular chondrocyte pellets after suramin treatment for 14 days compared with control (*P<0.05 for suramin treatment, two-way analysis of variance (ANOVA); P=0.14 and 0.07 for healthy and osteoarthritis (OA) cells, respectively, by Sidak test). (C) Quantitative assessment of immunohistochemical stainings demonstrating increased TIMP3 (*P<0.05 for suramin treatment, two-way ANOVA; P=0.5 and 0.03 for healthy and OA cells, respectively, by Sidak test) and reduced (D) VDIPEN (*P<0.01 for suramin treatment, two-way ANOVA; P=0.02 and 0.03 for healthy and OA cells, respectively, by Sidak test) and (E) NITEGE levels in suramin-treated human articular chondrocyte pellets relative to controls (*P<0.01 for suramin treatment, two-way ANOVA; P=0.005 and 0.31 for healthy and OA cells, respectively, by Sidak test). Individual data points are the means of three technical replicates per experiment.
Discussion
This study demonstrates that suramin has chondroprotective properties in a mouse model of cartilage damage and augments proteoglycan content in human articular chondrocyte pellet cultures by increasing the levels and biological activity of TIMP3 in the ECM. This results in an enhanced inhibitory effect on the activity of matrix degrading enzymes such as MMPs and ADAMTSs and in a reduction of cartilage breakdown.
Suramin is a highly sulfonated compound that was earlier used to extract TIMP3 from rat uterus tissue.25 TIMP3 is the only member among the TIMP family that can inhibit both ADAMTs and MMPs. Moreover, TIMP3 has the unique ability to bind to the ECM in cartilage, in contrast to the other TIMPs (TIMP1, TIMP2 and TIMP4) that diffuse freely within the matrix.27 The N-terminus of TIMP3 contains a highly basic region that is known to interact with negatively charged sulfated ECM components.25 26 These unique characteristics position TIMP3 as the main inhibitor of the ECM turnover.25 37 Thus, identification of pharmacological drugs that enhance TIMP3 levels in the extracellular environment is clinically promising to prevent ECM degradation.25 Here, we demonstrate that accumulation of TIMP3 in the ECM is the major underlying mechanism of suramin’s chondroprotective effects. Hence, a neutralising antibody targeting TIMP3 blocks the effect of suramin on proteoglycan accumulation in the micromass model. TIMP3 immunoprecipitation with a C-terminal-directed TIMP3 antibody showed the endogenous interaction between TIMP3 and suramin. Therefore, the interaction between suramin and TIMP3 may be likely localised at the N-terminal side of TIMP3. Recent work suggested that the interaction of suramin and TIMP3 inhibited endocytosis of TIMP3 mediated by its interaction with the endocytic scavenger receptor, low-density lipoprotein receptor-related protein 1.27 Although we cannot exclude additional effects of suramin on articular chondrocytes,28–30 our data and the work of Chanalaris suggest that interaction with TIMP3 is the main mechanism involved.
Studies in mice lacking TIMP3 showed spontaneous increased metalloproteinase activity and age-dependent cartilage degeneration, similar to changes seen in patients with osteoarthritis.38 Several diseases, such as atherosclerosis,39 cancer40 and osteoarthritis,41 are associated with reduced tissue levels of TIMP3. In human osteoarthritis cartilage, TIMP3 mRNA levels are not significantly changed, while TIMP3 protein levels are reduced.41 42 The addition of exogenous TIMP3 blocks cartilage degeneration in bovine nasal and porcine articular cartilage explant cultures.43 Additionally, TIMP3 injection blocks cartilage breakdown in a rat surgical model of osteoarthritis.44 Interestingly, our data therefore identify suramin as a new pharmacological modulator of TIMP3’s functional levels in cartilage.
We studied suramin in vivo using the papain model. This model results in direct cartilage damage and is therefore translationally relevant in the context of high-impact trauma and cartilage lesions. On impact trauma or disruption of the cartilage integrity, tissue destructive enzymes are activated.3 45 Although the subsequent development of osteoarthritis is a slow and steady process, we speculate that local treatment with suramin after cartilage damage may prevent further joint injury. Suramin is a market-authorised drug, and clinical pharmacology data are available. This means that in vitro and in vivo screening, chemical optimisation, toxicology, bulk manufacturing, formulation development and clinical development are completed. This leads to faster development time, reduced risks and costs in antiosteoarthritic pharmaceutical research.11 Intra-articular use of suramin appears to be the preferred way of administration. The pharmacokinetic profile for applying suramin systemically shows poor gastrointestinal tract absorption and several reversible adverse reactions.13 20 Intra-peritoneal injection of 1 mg suramin did not produce any beneficial effect to the proteoglycan content in the articular cartilage (data not shown). Most likely, the tissue distribution of suramin may have been insufficient to build up adequate amounts in the articular cartilage. This delivery limitation is probably due to both the avascular cartilage tissue and the dense superficial layer of articular cartilage matrix.46 To solve inefficient delivery to the target and too rapid clearance of the drug, we applied suramin locally by intra-articular administration. Testing the effects of suramin in a slow model of osteoarthritis such as destabilisation of the medial meniscus currently is a challenge as repeated intra-articular injections of the drug result in repetitive joint damage thereby interfering with our ability to evaluate the development of osteoarthritis in such a model. Hence, future directions of this work could be to explore slow-acting drug formulations for targeting articular cartilage with suramin, for example, using nanomedicine or nanodevices that are being used for in vivo delivery of biomolecules or to explore suramin analogues with improved physicochemical properties.47–50
In conclusion, our study suggests that suramin, which increases the TIMP3 amount and activity in the extracellular environment of the articular cartilage, would be a potential pharmacological drug to prevent progressive cartilage damage, in particular after cartilage trauma.
Acknowledgments
We are grateful to the traumatology and orthopaedic surgeons willing to contribute samples (A Sermon, JP Simon and S Nys) as well as the nursing staff of the surgical theater (in particular M Penninckx).
References
Footnotes
Contributors All authors contributed to the planning of the study, the design of the experiments and the analysis of the data. LAG, MK and JVD performed experiments. SM, GK, GO, RJL and FC supervised experiments. LAG, SM, RJL and FC wrote the manuscript. All authors provided input and suggestions and approved the final version of text.
Funding This work was supported by grant G.0A06.13 from the Flanders Research Foundation (FWO Vlaanderen). SM and FC were the recipients of Marie-Curie IEF Fellowships. The nano-CT images have been generated on the X-ray CT facility of the Department of Development and Regeneration of the KU Leuven, financed by the Hercules Foundation (project AKUL/13/47: Nano CT and Bioreactor CT for a better understanding of the dynamics of 3D tissue formation in regenerative medicine: from clustered cells toward organised multi-tissue system and whole organs).
Competing interests None declared.
Ethics approval UZ Leuven Ethical Committee.
Provenance and peer review Not commissioned; externally peer reviewed.