Article Text

Abstract
Epidemiological findings suggest a potential role for anti-citrullinated protein antibodies (ACPAs) in rheumatoid arthritis (RA) pathogenesis. ACPA-positive RA is associated with unique genetical and environmental risk factors, in contrast to seronegative RA. ACPA-positive healthy individuals are at risk of developing RA and can develop joint pain and bone loss already before disease onset. ACPA injection triggered bone loss and pain-like behaviour in mice and, in the presence of additional arthritis inducers, exacerbated joint inflammation. In cell culture experiments, ACPAs could bind to and modulate a variety of cellular targets, such as macrophages, osteoclasts, synovial fibroblasts, neutrophil granulocytes, mast cells, dendritic cells and platelets, further underlying a potential role for these autoantibodies in triggering pathogenic pathways and providing clues for their mechanisms of action. Patient-derived ACPA clones have been characterised by unique cellular effects and multiple ways to act on the target cells. ACPAs might directly induce stimulatory signals by ligating key citrullinated cell surface molecules or, alternatively, act as immune complexes on Fc receptors and potentially other molecules that recognise carbohydrate moieties. On the contrary to experimentally manufactured ACPA clones, patient-derived ACPAs are highly promiscuous and cross-reactive, suggesting a simultaneous binding to a range of functionally relevant and irrelevant targets. Moreover, several ACPA clones recognise carbamylated or acetylated targets as well. These features complicate the identification and description of ACPA-induced pathogenic mechanisms. In the current review, we summarise recent data on the functional properties of patient-derived ACPAs and present mechanistic models on how these antibodies might contribute to RA pathogenesis.
- anti-citrullinated protein antibodies
- arthritis
- rheumatoid
- autoimmunity
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Patient-derived anti-citrullinated protein antibodies (ACPAs) could modulate cells in culture and trigger pain and bone loss in mice.
In murine models, ACPAs exacerbated arthritis development in the presence of other inflammatory signals.
ACPAs might modulate cellular functions by directly ligating key citrullinated cell surface antigens or as part of immune complexes.
Despite their highly poly-specific and cross-reactive nature, different ACPA clones might have unique cellular targets and pathological roles.
Rheumatoid arthritis (RA) is a common autoimmune disease affecting close to 1% of the total population and having a heterogeneous clinical picture consisting of at least two different subsets with distinct genetic and environmental determinants. The main subset, comprising a majority of all cases, is the antibody-positive (or seropositive) RA and is characterised by the presence of antibodies reactive with the Fc-part of IgG (rheumatoid factor (RF)) and antibodies reactive to antigens that have been subjected to different post-translational modifications, mainly by citrullination (anti-citrullinated protein antibodies (ACPAs)) but also other modifications (then termed anti-modified protein antibodies (AMPAs)). Autoimmunity and specifically ACPAs are triggered before the onset of joint inflammation in susceptible individuals carrying certain genetic variants in MHC class II genes and exposed to different environmental challenges.1 It has been suggested that this autoimmunity might have direct pathogenic effects. The current review will address the potential pathogenic roles of ACPAs in human disease and experimental models.
Development of ACPAs in RA
ACPAs are a heterogenous group of antibodies defined by their capacities to recognise post-translationally modified citrullinated proteins/peptides but not their native counterparts. In the clinical context, these antibodies are measured by the anti-cyclic citrullinated peptide (anti-CCP) assay. Whereas this assay captures most of the ACPAs, there is a high heterogeneity in reactivity to distinct citrullinated proteins/peptides. More recently, cross-reactivity with other post-translational modifications has also been described with some ACPAs recognising acetylated and carbamylated proteins/peptides.2
ACPAs are present in 1%–3% of the general population, that is, among individuals without rheumatic complaints. They confer an increased risk for RA but the RA prevalence among asymptomatic individuals is low, making it conceivable that not all ACPA-positive individuals will end up developing the disease. Mechanisms responsible for tolerance breaking against citrullinated proteins and the development of ACPAs are still incompletely elucidated, but mucosal challenges by environmental pollutants, such as smoking, have been suggested to play an important role. Smoking could trigger immune activation in the lungs, which might lead to the production of certain autoantibody types, such as RF and ACPA, and occasional formation of germinal center-like structures in seropositive individuals who have not yet developed joint inflammation or in patients having a recent onset of seropositive RA. Smoking might also upregulate peptidyl-arginine deiminase (PAD) expression and activation in the airways and, consequently, increase the expression of citrullinated neoantigens.1
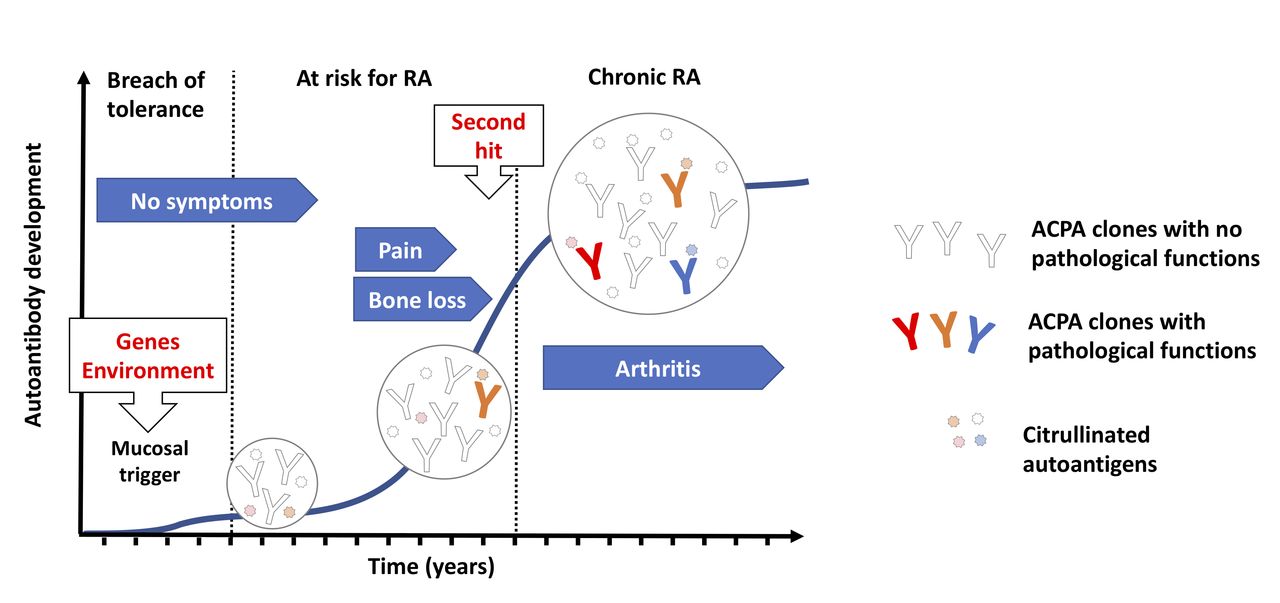
While the breach of immunological tolerance at mucosal sites might lead to autoimmunity, it does not fully account for disease development, as ACPAs could exists in asymptomatic individuals for a long time and not all ACPA-positive individuals develop arthritis. It is believed that other immune events are necessary for a boost of the ACPA response that will eventually lead to development of pathogenic autoimmunity. Interestingly, the major genetic risk factor for RA, encoded by the HLA-Shared epitope (SE) alleles, appears to be more important in the transition from autoimmunity to established disease rather than generation of autoimmunity itself.3 As such it is possible that the HLA-SE molecules boost the ACPA response towards a more pathogenic phenotype potentially through antigen presentation to T cells that will provide help to B cells recognising citrullinated antigens, while the initial breach of tolerance to citrullinated peptides occurs in an HLA-SE independent manner. Typically, seropositive individuals not developing RA have low levels of ACPAs with reactivity limited to few citrullinated epitopes. In contrast, an increase in isotype diversity, autoantibody levels and the number of recognised citrullinated epitopes are seen in seropositive individuals who develop RA, typically with a peak observed just before disease onset4 (see figure 1). More recently, another feature of the ACPAs, the abundance of variable domain glycans, has been suggested to play a role in ACPA maturation and repertoire expansion based on the finding that first-degree relatives of RA patients show an extensive variable domain glycosylation that is associated with future development of RA.5 However, the exact contribution to disease progression should be interpreted with caution as far as the abundance of variable domain glycans in ACPAs could be detected as long as 15 years before disease onset.6 Besides changes in variable region glycosylation, a decrease in the Fc-galactosylation of the IgG ACPAs, potentially affecting ACPAs interaction with the complement system and the Fc receptors, has been reported to occur during progression to arthritis.7 8 This suggest a causal relationship to the onset of joint inflammation, but further investigations to confirm the relevance of this finding are needed.
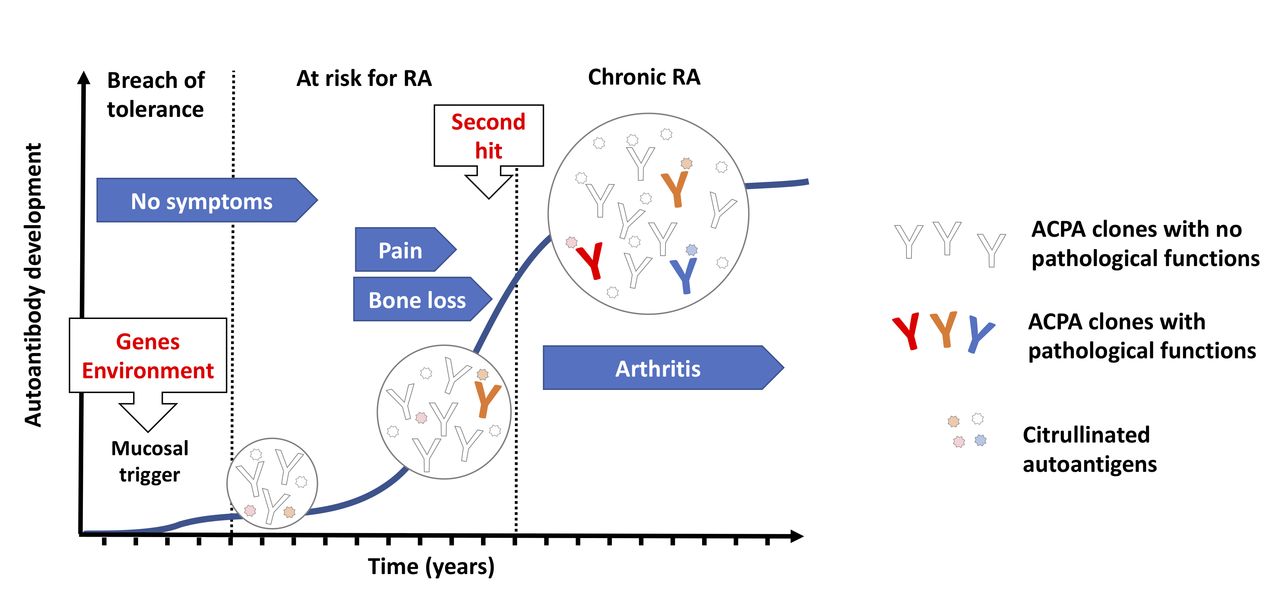
Evolution of ACPA reactivities during RA progression. Humoral autoimmunity against citrullinated autoantigens can be triggered in response to environmental challenges at mucosal sites (such as cigarette smoking and bacterial infections) especially in individuals with a genetical background that is linked to RA susceptibility. Individuals carrying ACPAs in the circulation can remain disease free for long periods, suggesting that the initial antibody repertoire may not obviously trigger pathogenic pathways. Nevertheless, ACPAs in patients with RA typically accumulate very high number of mutations suggesting a progressive evolution of the ACPA repertoire, via somatic hypermutation and antigen-driven selection, which can ultimately lead to novel fine specificities that enable the targeting of key signalling molecules on cells that are characterised by a steady state protein citrullination. ACPAs might thus trigger certain pathogenic signals already before disease onset, such as pain and bone erosion, and potentially facilitate arthritis development in response to additional arthritogenic stimuli representing a ‘second hit’ for arthritis development. RA onset is associated with an increase in ACPA levels and a further expansion of the antibody diversity. Inflammation boosts protein citrullination, which can lead to increased immune complex formation and the appearance of novel autoantigens, enabling further types of pathogenic ACPA signals. ACPAs, anti-citrullinated protein antibodies; RA, rheumatoid arthritis.
Changes in the ACPA structure and repertoire are paralleled by changes in the clinical picture, most often in those individuals who later develop RA, with an early development of musculoskeletal symptoms (such as pain and fatigue) and also signs of bone loss9 and subclinical inflammation,10 culminating in the development of clinical detectable arthritis (see figure 1). Once these individuals reach RA diagnosis, the ACPA repertoire remains relatively stable over time with small changes in the antibody levels and only occasional seroconversion. Nonetheless, the presence of ACPAs in established disease is an important prognostic factor associated with worse clinical and therapeutic outcomes, such as increased bone destruction, comorbidities and mortality as well as therapeutic failure (for review see the study by de Brito Rocha et al11). Taken together, the RA specificity, the early development of the ACPA response and its prognostic value in both individuals at risk and those already having RA suggest that ACPAs are involved in disease pathogenesis. Several lines of evidence coming from in vitro experiments in different cellular systems and in vivo animal models that support this hypothesis are currently available, with some still unsolved issues and controversies.
Pathogenic effects of ACPAs
Macrophage activation
Macrophages are important regulators of the chronic synovial inflammation in RA. These cells have an activated phenotype, express high levels of Fcγ receptors and are the major source of the proinflammatory cytokine tumour necrosis factor-α (TNF-α). As such, macrophages have been the cell type most studied in conjunction with potential pathogenic effects of ACPAs. Originally, polyclonal ACPAs have been shown to activate macrophages and promote production of proinflammatory cytokines through an immune-complex (IC)-mediated mechanism dependent on the Fcγ receptors and activation of the TLR4-MyD88 pathway.12–15 Incorporation of RF in the ACPA-IC augmented this effect.16 17 Later on, monoclonal ACPAs generated from either the blood or the synovial tissue of patients with RA were also shown to activate macrophages through a similar IC-mediated mechanism.18 19 Notably, all these experiments have been carried out using plate-bound IC, making it challenging to extent these findings to in vivo situations where the structure and availability of the ACPA-ICs could be different. Another group has proposed an alternative mechanism showing that polyclonal ACPAs in the absence of preformed ICs are still able to activate macrophage-like cells and induce TNF-α production through activation of the extracellular signal-regulated kinase, c-jun-N-terminal kinase and nuclear factor-κB (NF-κB) pathways.20 21 ACPAs, in the absence of preformed ICs, not only activated macrophages but also promoted their polarisation towards an M1 proinflammatory phenotype through an interferon regulatory factor 5-dependent mechanism.22 More recently, it has been shown that polyclonal ACPAs (again in the absence of preformed ICs) can upregulate the interaction between CD147 and integrinβ1 in macrophages followed by the activation of the protein kinase B (Akt)/NF-κB signalling pathway and induction of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) and pro-IL-1β expression.23
Osteoclast activation and bone loss
Osteoclasts are highly specialised cells derived from haematopoietic precursors of the myeloid lineage and responsible for bone resorption. Bone loss due to increased bone resorption and impaired bone formation occurs in RA, especially in longstanding and treatment-resistant disease, with development of joint erosions, periarticular bone loss and systemic osteopenia. While the classical view is that inflammation is a major determinant of these changes, increasing evidence shows a strong association between ACPA seropositivity and bone loss, not necessarily coupled to inflammation. As such, ACPAs associate with bone loss in both individuals at risk but not yet having disease as well as in patients with newly diagnosed untreated RA (for review see the study by Kocijan et al9). Interestingly, a study in mice showed that immunisation with citrullinated type II mouse collagen led to increased ACPA levels, inversely correlating with the bone quantity and quality, but uncoupled from the degree of inflammation.24 These findings suggest a direct effect of ACPAs on the bone, independently of the inflammation and chronic disease burden.
Originally, it was shown that antigen-specific binding of citrullinated vimentin by polyclonal anti-modified citrullinated vimentin antibodies promoted osteoclasts activation and differentiation both in vitro and in vivo when the antibodies were injected into Rag1 knockout25 or wild-type mice,26 potentially through a TNF-α-mediated mechanism. We later confirmed that polyclonal ACPA preparations promoted osteoclast development in vitro, using affinity purified anti-CCP2 antibody fraction, and we described that the antibodies acted via a peptidyl arginine deiminase and IL-8-dependent mechanism.27 We simultaneously reported the effect of several RA-derived monoclonal antibodies, at that time believed to be ACPAs, of which some were able to promote osteoclast activation and bone loss. However, continuous work and refined characterisation of some of these antibodies revealed that they lack specific binding to citrullinated peptides in surface plasmon resonance, as described in a correction note to Annals of the Rheumatic Diseases. Therefore, the results reported for these monoclonal antibodies cannot be attributed to reactivity against citrullinated proteins and/or peptides, but are due to other yet unknown mechanisms.28 Reactivity to other post-translational modifications or to other osteoclast-specific autoantigens and/or an FcR-dependent mechanism might partially explain the observed effects. Importantly however, more recently by using the next generation of well-characterised monoclonal ACPAs we have shown that indeed certain monoclonal ACPA can induce osteoclastogenesis.29 30 Taken together, the data implying a direct antigen-specific effect of ACPAs on osteoclasts, while appealing in the light of the clinical association between ACPA and bone destruction, needs further validation and characterisation.
Besides the ligation of cell surface molecules, antibodies can engage Fc receptors in the form of immune complexes. Importantly, osteoclasts express several Fcγ receptors and IgG containing ICs can activate osteoclasts and increase bone loss independently of the antigen specificity.31–33 The osteoclast activating effect of IgGs appears to be dependent on the Fc glycosylation status, IgG isotype and the different distribution of Fcγ receptors in physiological compared with inflammatory conditions.34 If the ACPA containing ICs exert similar Fc-dependent effects, it still remains to be demonstrated.
Modulation of synovial fibroblasts
Synovial fibroblasts constitute an inflammatory stroma in RA joints that perpetuate immune activation by releasing cytokines and chemoattractants and that is able to erode the cartilage. Importantly, synovial fibroblasts can upregulate PAD enzyme expression in an inducible manner, in response to cellular stress such as hypoxia or serum starvation, and the cytokine IL-8.35 36 The increase of PAD expression was accompanied by ACPA binding to the cell surface. In line with the inducible nature of ACPA sensitivity, ACPAs could bind to fibroblasts in the inflamed synovia of patients with RA but not in healthy, non-inflamed joints.35 ACPA binding affected cell adhesion and increased cell mobility in wound-healing assays, without a major effect on cell viability or the secretion of a set of cytokines and matrix metalloproteinases.35 It has remained to be understood how ACPA-mediated changes in fibroblast mobility could contribute to RA pathogenesis or whether ACPAs could also modulate further functions in these cells.
Induction of NETosis and NET-derived IC formation
Neutrophil granulocytes are innate immune cells with potential roles in the loss of immune tolerance and in promoting synovial inflammation during RA. Neutrophil activation can lead the extrusion of cellular DNA and protein complexes that form neutrophil extracellular traps (NETs) with antimicrobial properties, through a form of cell death coined NETosis. While originally it has been reported that ACPAs might stimulate NETosis,37 38 a more recent study failed to confirm this observation.39 ACPAs, on the contrary, might form ICs with citrullinated antigens released during NETosis. ACPAs isolated from the inflamed synovium frequently showed a high reactivity to modified proteins exposed in the NETs, and a range of ACPA clones could bind to NETs induced by a variety of stimuli in vitro.40–42 It is also important to note that neutrophils seemed to be primed for undergoing NETosis in patients with RA,37–40 suggesting that NET-derived autoantigens might be more abundant in RA compared with healthy individuals. Moreover, several of the ACPA clones cross-react with homocitrullinated and acetylated epitopes, which can be present in activated neutrophils, suggesting a possibility of PAD-independent autoantigen production for ACPAs in NETosis.2 41
Pain induction
The clinical association between joint pain and ACPA responses in seropositive individuals that eventually develop arthritis has suggested a possible causal relationship. Indeed, systemic polyclonal ACPA injection in mice leads to pain-like behaviour in the absence of any sign of local or systemic inflammation.43 While this was originally reported for both polyclonal and monoclonal ACPAs, the results on monoclonal antibodies as published should be disregarded as far as it has been shown that these monoclonals were not true ACPAs. Interestingly, systemic administration of antibodies specific for collagen type II or cartilage oligometric matrix protein was reported to also induce changes in the pain-like behaviour in mice, through antigen recognition and immune complex formation in the absence of inflammation.44 In a similar way, local injection of IgG immune complexes into the joints evokes acute joint hypernociception without obvious concurrent joint inflammation through an Fc-mediated mechanism.45 Taken together, these findings might explain why joint pain predates inflammatory arthritis onset in RA but it still remains to be determined if the effect of ACPAs on pain behaviour is mediated through an FcR-dependent or FcR-independent mechanism.
Immunogenicity of citrullinated antigens and exacerbation of the arthritis by ACPAs
Intense efforts to link ACPAs to arthritis development in animal models have been ongoing since the recognition of these antibodies in the human disease, but the results have so far remained highly variable and to a certain degree contradictory. These differences could be explained by differences in mice strains, antigens used for immunisation or antibodies used for passive transfer, definition and scoring of joint inflammation as well as environmental characteristics in different laboratories.
ACPA formation and arthritis development have been originally described following immunisation with citrullinated fibrinogen and adjuvant (complete Freund’s adjuvant (CFA)) in DR4 transgenic mice,46 47 but this finding was not confirmed in an independent study.48 Similarly, immunisation with a homocitrullinated peptide gave rise to both anti-homocitrullinated and anti-citrullinated target-specific responses in DR4 transgenic mice, suggesting cross-reactivity dependent on the SE, but it did not lead to arthritis development.49 However, immunisation of DR4 transgenic mouse with either citrullinated or native Porphyromonas gingivalis (P. gingivalis)-derived or human enolase led to formation of ACPAs and development of paw swelling/arthritis.50
Besides these few studies in DR4 transgenic mice, a large majority of reports have addressed the immunogenicity and arthritogenicity of citrullinated peptides/proteins in non-humanised animal models. Several studies have shown that different citrullinated peptides/proteins in the presence of CFA are able to brake tolerance and, in some cases, to induce and/or aggravate arthritis. In rats for example, immunisation with citrullinated fibrinogen51 or citrullinated albumin52 induced ACPAs but no clinical or histological signs of arthritis. Moreover, immunisation with citrullinated fibrinogen was not able to aggravate a pre-existing joint inflammation.51 In contrast, immunisation with either citrullinated histone 2B,53 citrullinated fibrinogen54 or citrullinated BiP55 gave rise to ACPA response in mice and exacerbated a pre-existing joint inflammation, without inducing arthritis in itself. In a ‘hapten/carrier’ model, T-cell immunisation against PAD (the carrier) triggered the development of antibodies to citrullinated fibrinogen peptides (in this case the hapten), but still no arthritis, as demonstrated in CH3 mice carrying an IEBk chain, which is highly homologue with the β1 chain HLA-DRB1*04:01.56 While these studies have been performed using CFA, two studies investigated the effect of immunisation with citrullinated type II mouse collagen in the absence of adjuvant. They reported joint inflammation and bone loss as well as an increase in the ACPA levels, inversely correlating with the bone quantity and quality.24 57 In another model, infection with P. gingivalis expressing a bacterial PAD enzyme has been shown to trigger arthritis and an ACPA response that correlated with local and systemic bone loss58 and to exacerbate arthritis development in ZAP-70 mutant (SKG) mice with increased production of ACPA.59
Importantly, citrullinated peptides could also downmodulate disease activity if introduced in the absence of adjuvant and before the onset of inflammation, as shown in the murine collagen-induced arthritis model.60 Similarly, in a rat adjuvant-induced arthritis model, subcutaneous injection of citrullinated peptides very early after disease initiation could reduce disease severity, likely through an increase in regulatory T cell/TH17 cell ratio and an effect on T cell apoptosis.61 These studies further emphasised the pathogenic nature of the autoimmunity against citrullinated proteins and also suggested the possibility of tolerance induction through the early rewiring of autoreactive T cells.
The real challenge in demonstrating a direct pathogenic effect of ACPAs on arthritis induction has been and is still the failure to induce disease by passive transfer of the antibodies. However, there are a handful of studies showing that ACPAs, while unable to induce arthritis by themselves, might still exacerbate pre-existing joint inflammation. As such, transfer of monoclonal antibodies specific to citrullinated fibrinogen was able to enhance arthritis when co-administered with a submaximal dose of anti-collagen antibodies.60 Similarly, human monoclonal ACPAs cloned from B-cells recognising filaggrin and CEP-1 enhanced the inflammation induced by joint injection of lipopolysaccharide (LPS), while not inducing arthritis when used alone.62 Murine anti-citrullinated collagen antibodies were able to induce a mild arthritis, exacerbated by either LPS boosting or the co-administration of other pathogenic anti-collagen antibodies.63 64 More recently, a human monoclonal ACPA with main reactivity against citrullinated fibrinogen was able to exacerbate experimental arthritis when injected into SKG mice along with Laminaria digitate.65 Taken together, these data suggested the need for additional triggers (second hits) to set the stage for the arthritogenic effects of the ACPAs (figure 1).
Other ACPA effects
ACPAs have been suggested to play a role in mast cell activation through a synergistic effect between ACPA containing ICs and a concomitant toll-like receptor stimulation.66 Similar to other antibodies, ACPAs can promote complement activation through both classical and alternative pathways, as shown by in vitro complement activation using plate-coated polyclonal anti-CCP antibodies isolated from the serum of patients with RA67 and plate-coated polyclonal ACPA-IC in the presence of IgA and IgM RF.68 We have recently shown that ACPAs can directly stimulate the trans-differentiation of dendritic cells (both in vitro generated and conventional circulating CD1c+ DCs) into osteoclasts through a PAD and IL-8-dependent mechanism.30 ACPA has been also shown to induce platelet activation through an FcγRIIa-mediated pathway.69
Cellular mechanisms responsible for the pathogenic effect of ACPAs
ACPA binding to cellular targets
The functional studies using different ACPA preparations have so far suggested two major mechanisms behind the action of the antibodies on their target cells. First, ACPAs might trigger signalling processes by ligating key citrullinated proteins on the cell surface (figure 2A), which could be the case in osteoclast and fibroblast cultures, where both ACPAs and ACPA Fab2 fragments acted similarly on the cells.30 35 Alternatively, ACPAs can form immune complexes and activate Fc receptors-positive cells (figure 2B).13 15 53 RFs might enhance ACPA-mediated mechanisms, by crosslinking ACPA-antigen complexes and thereby enhancing the signals triggered by direct ACPA binding to the cell surface or, alternatively, by increasing the size of immune complexes and thereby promoting Fc receptors activation (figure 2C).16 35 These two pathways are not necessarily exclusive but might co-exist on the same cell type (figure 2D). Alternatively, ACPAs might act via one of these mechanisms only, depending on the presence or absence of key citrullinated antigens and FcRs on the cell surface. Synovial fibroblasts, for example, lack IgG binding Fc receptors but reacted to ACPAs, suggesting a stimulatory effect of the antigen binding itself, whereas osteoclasts might react via both FcR-dependent and FcR-independent mechanisms.31 35 Physiological immune complex formation is bypassed experimentally in several studies by heat aggregation of the studied antibodies (figure 2E).
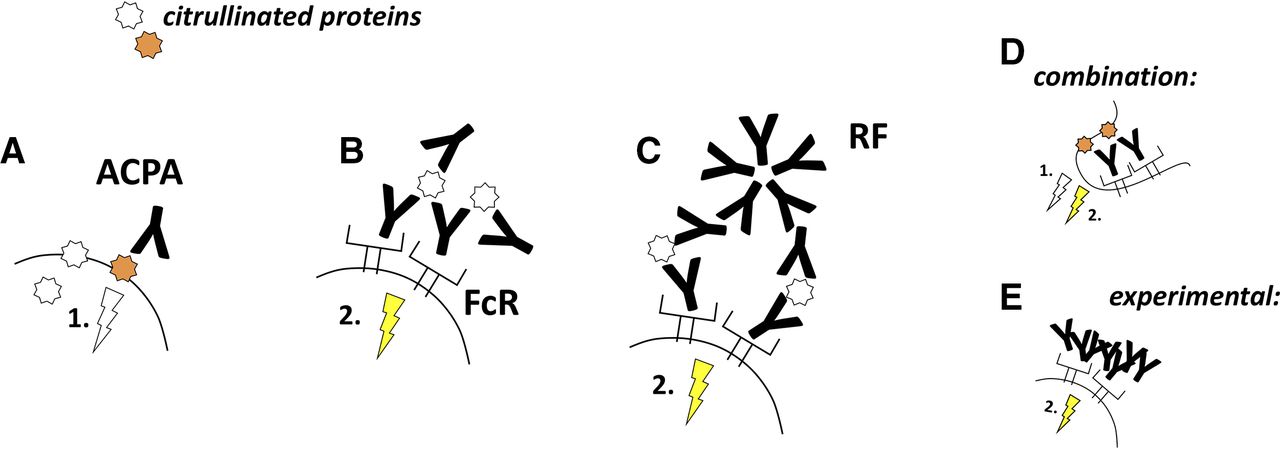
ACPAs modulate cellular function by binding to cell surface proteins or by forming immune complexes. (A) ACPA binding to key citrullinated proteins on the cell surface might directly initiate signalling processes in the cells. (B) Alternatively, ACPAs can form immune complexes and activate FcRs. (C) Rheumatoid factor antibodies could enhance both mechanisms by crosslinking ACPA-antigen complexes and thereby increasing direct or FcR-mediated ACPA signals (the latter is illustrated). (D) Different ACPA-mediated pathways might concomitantly occur in the same cells that express functional ACPA targets and FcRs. (E) Autoantibody effects are frequently studied by using heat-aggregated immune complexes, bypassing the role of the antigens in the antibody-induced signalling. Such models have provided some important information but, at the same time, lack the ability to embrace other aspects such as the direct cell stimulation by ACPA ligation of cell surface proteins or whether the antibodies can form FcR stimulating ICs. ACPAs, anti-citrullinated protein antibodies.
It is noteworthy that the lack of citrulline reactivity in case of some of the antibody clones used in the early ACPA functional studies triggered concerns about the specificity of the ACPA effects in other studies as well.28 Protein aggregation and minute microbial contaminations could obviously affect cellular responses to antibodies, which highlight the importance of the vigorous quality testing of ACPA preparations before the evaluation of any functional activity.70 Importantly, in more recent functional studies, ACPA and control antibody preparations were characterised by similarly low aggregation levels and a lack of measurable LPS contamination, which was evaluated by both the limulus amebocyte lysate test and a TLR4-dependent macrophage bioassay.30 35
Poly-specific ACPAs mediate specific functions
ACPA clones that have been so far isolated from patients with RA and characterised in detail all have a relatively high level of polyspecificity, in some cases showing reactivity against several thousands of unique citrullinated epitopes.29 62 It is an intriguing question whether and how such highly promiscuous antibodies could have specific roles in disease development. ACPA clones very often recognise a core consensus motif in the different epitopes, where citrulline is adjacent to glycine (ie, an amino acid without side chain), suggesting the importance for a proper accessibility of citrulline for antibody binding. However, a recent comprehensive report revealed that further amino acids in the vicinity of the citrulline can influence the binding of different ACPA clones both positively and negatively, leading to a somewhat unique fine-specificity pattern for the individual clones.2 Nevertheless, even ACPA clones with largely overlapping target repertoire are in reality characterised by highly variable binding affinities to different target sequences (potentially up to 10-fold to 100-fold differences).2 29 Such variety of binding affinities suggests a certain level of specificity even for polyreactive antibodies in physiological circumstances, where different clones would prioritise the highest affinity targets from the available epitopes (figure 3A). In addition, non-overlapping target epitopes have been identified for all tested ACPA clones, implying the possibility of clone-specific pathogenic features, even for otherwise promiscuous antibodies, as suggested by distinct cellular effect of different ACPAs clones.29 30 35 ACPAs therefore seem to incorporate both specificity and promiscuity into their features by binding a few selected epitopes with high specificity, while binding to other epitopes with either relatively higher or lower affinity when compared with other clones. Such hierarchy of the recognised epitopes can determine the functional capacities of the individual clones, that is, ACPAs can stimulate target cells by preferentially binding to key functional epitopes on the cell surface, whereas recognising functionally irrelevant targets might compete with this effect (figure 3B). Binding to functionally irrelevant antigens would not lead to any effect, even in the case of highly specific autoantibodies. Irrespectively, promiscuous binding to a diverse range of citrullinated proteins can also bear functional significance as it might facilitate immune complex formation (figure 3C).

{kind=link}
{kind=link}
{kind=link}
Poly-specific ACPAs with specific functions. (A) ACPAs isolated from patients with rheumatoid arthritis typically recognise a very high number of citrullinated antigens in binding assays. However, each antibody is characterised by different affinities towards the individual antigens, which leads to their preferential binding to the highest affinity targets under physiological conditions. (B) ACPAs can directly stimulate cells by binding to a key functional target on the cell surface. Highly specific (dark blue) or poly-specific/promiscuous (light blue) autoantibodies can trigger similar activation signals, if the ligation of such surface molecules is sufficiently strong. Diversion of a promiscuous antibody to irrelevant targets could, however, decrease signal strength. Ligation of irrelevant surface antigens, either by highly specific or promiscuous antibodies will not trigger activation signals. (C) Promiscuity of the autoantibodies can facilitate immune complex formation and FcR binding. ACPAs, anti-citrullinated protein antibodies.
Similar action of different AMPA types
As mentioned above, besides ACPAs, antibodies directed against other post-translational modifications (generated by very different mechanisms than citrullination) such as carbamylation, acetylation or malondialdehyde (MDA) modifications, are present in RA.71 Interestingly, approximately half of the ACPA clones obtained from patients with RA reacted with carbamylated antigens and some of these also showed reactivity to acetylated peptides.2 29 41 An explanation for such relatively frequent cross-reactivity might be the similar electron density distributions at the key recognised residues as, at least some of the tested ACPA clones retained their binding capacity after replacing citrulline to homocitrulline or acetylated-lysine in the same peptide antigen.2 While not cross-reactive with citrullinated antigens, anti-MDA antibodies have been shown to activate osteoclast similar to ACPAs, indicating that similar pathways can be triggered by targeting antigens that are generated via different protein modifications.29 30 35 72 Understanding which types of protein modifications are key for certain autoantibody effects is complicated due to several reasons. First, in spite of the diverse mechanisms, such protein modifications often co-exist, for example, cigarette smoke can trigger citrullination and carbamylation and inflammation stimulates many types of post-translational modifications.71 73–75 Further, osteoclasts are characterised by steady state protein citrullination as well as MDA modifications, potentially even at overlapping protein residues.72 Citrullination of actin 62–68 peptide, for example, has been described in osteoclasts cultures and in the inflamed synovium, where the same sequence was also identified with MDA modification.30 72 76 Activated neutrophils might also generate citrullinated, carbamylated, acetylated as well as MDA-modified proteins and, through NETosis expose these for autoantibodies targeting the modified self-proteins.41 71 Different types of protein modifications can thus co-exist in the same tissue environment, in the same cells, in the same proteins and occasionally even at the same peptide epitope, which provides a likely explanation for the co-existence of autoantibodies targeting different post-translational modifications in the same disease and also for their similar functional capacities.
Conclusions and future perspectives
ACPAs are important disease biomarkers in RA with several suggested pathogenic effects related to the classical effector functions of antibodies and potentially to some ACPA-specific traits. Experimental data describing that different ACPA clones act on different cell types, which displayed citrullinated antigens either spontaneously or in an inducible manner, suggest a scenario where ACPAs might trigger a chain of separate pathological pathways by acting on early and late cellular targets during RA progression. Osteoclasts and their precursors characterised by steady state protein citrullination might be targeted by ACPAs already before disease onset, in the absence of an inflammatory milieu, contributing to the earliest pathological signs of RA progression, such as arthralgia and microscopical erosions of the bones. In contrast, synovial fibroblasts and potentially other cells, such as neutrophils, need to be activated for the binding of ACPAs, suggesting that these cells might be targeted later, in the already inflamed joints. It is also possible, on the other hand, that ACPA specificities required for targeting different cell types appear rather stochastically in the course of the disease, leading to a large individual variability in the cellular effects of the ACPA repertoire. ACPA clones that stimulate osteoclasts or fibroblasts, for example, might appear early or late in the disease or could be completely absent in different individuals. ACPAs, in this case, would trigger individual mosaics of different pathological pathways, playing more or less significance in the overall disease progression in different patients. Inevitably, to envisage the role of ACPAs in RA pathology, further studies are needed comparing different types of ACPA preparations obtained from different patients and from different stages of the disease, in the experimental models of pain, bone erosion and synovial inflammation.
References
Footnotes
Deceased Professor Anca Catrina Deceased on Feb 2021
Contributors All authors have contributed to the design, interpretation and writing of the current review.
Funding This project has received funding from FOREUM, Foundation for Research in Rheumatology, from the European Research Council (ERC) grant agreement CoG 2017 - 7722209_PREVENT RA, the EU/EFPIA Innovative Medicine Initiative grant agreement 777357_RTCure, the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse and Knut and Alice Wallenberg Foundation.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Data availability statement No additional data are available.